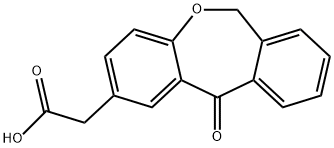
Isoxepac synthesis
- Product Name:Isoxepac
- CAS Number:55453-87-7
- Molecular formula:C16H12O4
- Molecular Weight:268.26
(1) condensation: prepare p-hydroxyphenylaceticacid 8-12 part, phthalide 6-12 part, sodium methylate 8-12 part by weight; Dissolve with DMAC and to add said sodium methylate behind said p-hydroxyphenylaceticacid and the phthalide; Be that 0.1-10Pa is heated to 80-170 ℃ of reaction 3-10h at pressure then; Regulate the pH value to 1-5,4-(2-carboxyl benzyloxy) toluylic acid is separated out in crystallization;
(2) cyclization: with Glacial acetic acid min. 99.5 dissolving step (1) gained 4-(2-carboxyl benzyloxy) toluylic acid, adding 3-52 weight part polyphosphoric acid again, is that 0.1-10Pa is heated to 30-100 ℃ of reaction 3-12h at pressure, and crystallisation by cooling gets the Isoxepac bullion then;
(3) purify: behind the said Isoxepac bullion of acetic acid ethyl dissolution, refining decolouring gets the Isoxepac product.
https://patents.google.com/patent/CN102838582A/en
![BENZENE ACETIC ACID, 4-[(2-CARBOXYPHENYL)METHOXY]](/CAS/GIF/55453-89-9.gif)
55453-89-9
137 suppliers
$20.00/1g
55453-87-7
348 suppliers
$5.00/25mg
Yield:55453-87-7 96.4%
Reaction Conditions:
Stage #1: 4-(2-carboxybenzyloxy) phenyl acetic acidwith trifluoroacetic anhydride in chlorobenzene at 20; for 4 h;
Stage #2: boron trifluoride diethyl etherate in chlorobenzene at -10 - 0; for 0.666667 h;
Steps:
1.5
The whole quantity of the 2-(4-carboxymethylphenoxymethyl)benzoic acid obtained in the above Step 4 and 200 ml of chlorobenzene were charged into a 500-ml four-necked flask, 75.0 g (0.3753 mol) of trifluoroacetic anhydride was added thereto, and the mixture was stirred at about 20°C for 4 hours. Subsequently, 2.3 g (0.0162 mol) of BF3-etherate complex was added dropwise thereto at -10 to 0°C over 10 minutes, then the mixture was stirred for 30 minutes. An aqueous phase was then separated, and the organic layer was washed with 200 ml of water. The washed organic layer was added to a solution of 7.2g of sodium hydroxide dissolved in 300 ml of water, and stirred for 30 minutes. An aqueous phase was then separated. To the separated aqueous layer, 2.0 g of activated carbon was added, stirred for 30 minutes, and filtered though a Buchner funnel to separate activated carbon. The activated carbon was washed on a Buchner funnel with 10 ml of water. A mixture of the filtrate and the cleaning solution was maintained at about 40°C, and a mixed solution of 11.3 g (0.1876 mol) of acetic acid and 50 ml of water was added dropwise thereto over 30 minutes. After the dropwise addition was completed, the mixture was cooled to 0 to 10°C, filtered through a Buchner funnel to collect crystals, and washed with 200 ml of water. The washed crystals were dried under reduced pressure to obtain 42.0 g of the title compound, (11-oxo-6,11-dihydrodibenz[b,e]oxepin-2-acetic acid). The yield (yield from 2-(4-carboxymethylphenoxymethyl)benzoic acid) was 96.4%, and the purity as measured by HPLC was 99.9%.(HPLC conditions) Column: Inertsil ODS-5 μm (4.6 mm ID x 15 cm)Mobile Phase: 0.02% trifluoroacetic acid aqueous solution/acetonitrile = 5/5 → 3/7 (30 minutes)Detection Wavelength: UV 254 nm(Physical Property Data) 1H NMR (400 MHz, DMSOd6) δ 3.63 (s, 2H), 5.30 (s, 2H), 7.07 (d, J = 8.0 Hz, 2H), 7.48 (t, J = 3.6 Hz, 1H), 7.55 (d-d, J = 7.9 Hz, 2H), 7.67 (t, J = 7.6 Hz, 1H), 7.78 (d, J = 7.6 Hz, 1H), 7.97 (d, J = 2.0 Hz, 1H)
References:
EP2072507,2009,A1 Location in patent:Page/Page column 10-11
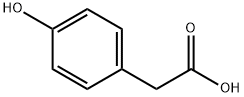
156-38-7
665 suppliers
$6.00/25g
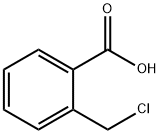
85888-81-9
148 suppliers
$13.00/250mg
55453-87-7
348 suppliers
$5.00/25mg
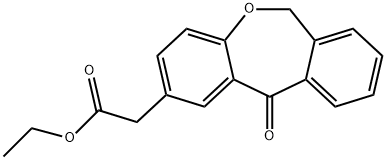
55453-90-2
12 suppliers
inquiry
55453-87-7
348 suppliers
$5.00/25mg
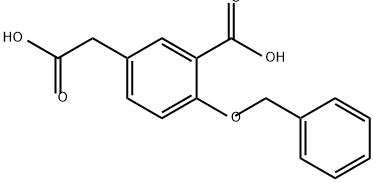
69031-39-6
0 suppliers
inquiry
55453-87-7
348 suppliers
$5.00/25mg

2417-73-4
207 suppliers
$84.00/5g
55453-87-7
348 suppliers
$5.00/25mg