| 94.2% |
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃; for 16h; |
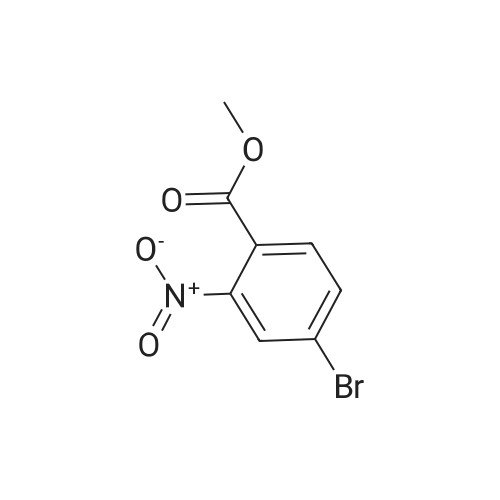
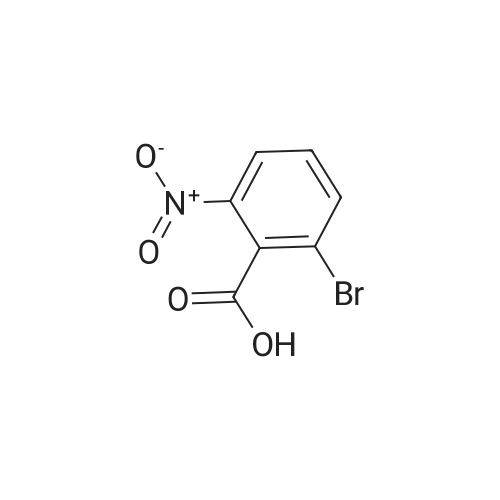
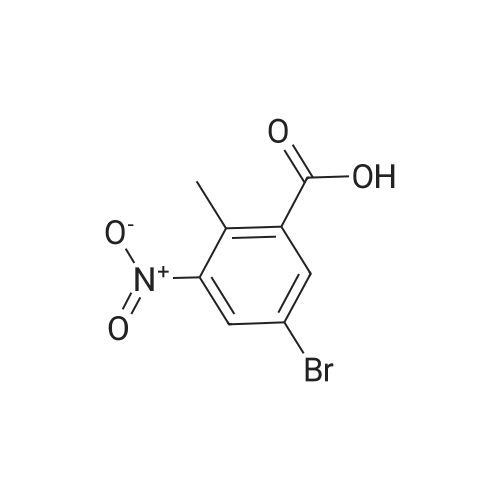
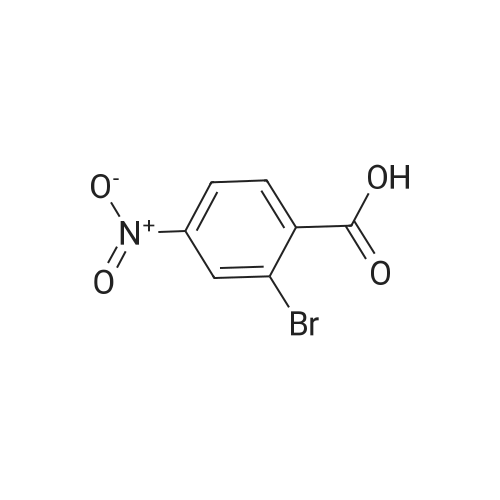
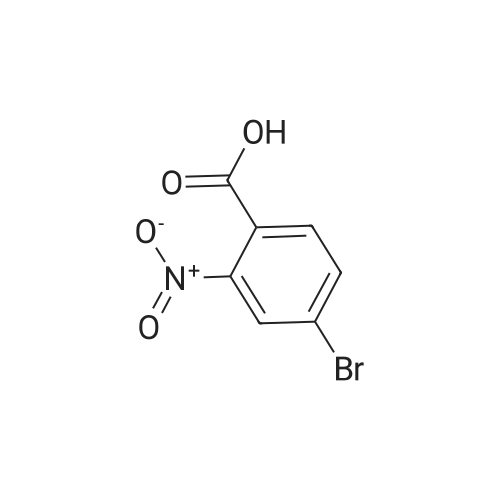
To a solution of compound 267a (5 g, 4.07 mmol), 4-bromo-2-nitrobenzoic acid in N, N-dimethylformamide (50 mL), cesium carbonate (13.5 g, 40.5 mmol) and methyl iodide (3.8 mL, 61 mmol).The mixture was stirred at room temperature for 16 hours, then the mixture was concentrated under reduced pressure to remove some solvents, water was added to the residue and extracted with ethyl acetate (160 mL × 2), and washed with saturated brine (60 mL × 3),After drying over anhydrous sodium sulfate, the filtrate was concentrated under reduced pressure, and the residue was purified by silica gel chromatography (petroleum ether: ethyl acetate = 91: 9) to obtain a white solid compound 267b, namely 4-bromo-2-nitrobenzene Methyl formate (4.98 g, 94.2% yield. |
| 90% |
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In DMF (N,N-dimethyl-formamide); at 0 - 20℃; |
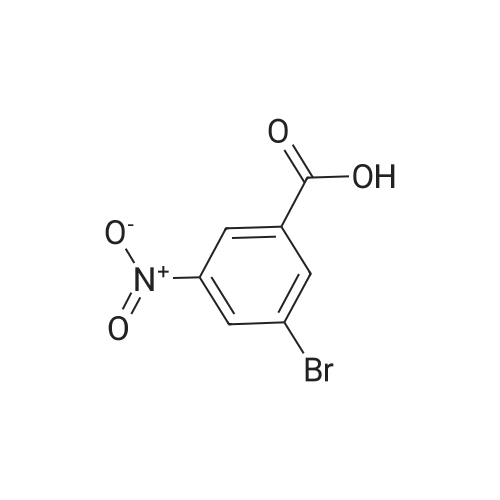
A. Methyl 2-amino-4-bromobenzoate To a stirred solution of 4-bromo-2-nitrobenzoic acid (EXAMPLE 5) (3.81 g, 15 mmol) in DMF (30 mL) at 0 C. was added 1,8-diazabicycloundecane (DBU) (10.3 mL, 75 mmol) followed by methyl iodide (4.67 mL, 75 mmol). The reaction mixture was stirred 15 min at 0 C. then allowed to warm to room temperature and stir overnight. The mixture was poured into water and extracted with EtOAc (2*). The combined organic extracts were washed with water (2*), dried (MgSO4), and concentrated in vacuo. The residue was purified by flash chromatography (hexanes/EtOAc) to afford methyl 4-bromo-2-nitrobenzoate as a pale yellow solid (3.52 g, 90%). |
| 90% |
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In N,N-dimethyl-formamide; at 0 - 20℃; |
B. Methyl 2-amino-4-bromobenzoate. ; To a stirred solution of 4-bromo-2-nitrobenzoic acid (3.81 g, 15 mmol) in DMF (30 mL) at 0 C. was added 1,8-diazabicycloundecane (DBU; 10.3 mL, 75 mmol) followed by Mel (4.67 mL, 75 mmol). The mixture was stirred for 15 min at 0 C., then was allowed to warm to rt and was stirred overnight. The mixture was poured into water and extracted with EtOAc (2×). The combined organic extracts were washed with water (2×), dried (MgSO4), and concentrated. The residue was purified by flash chromatography (hexanes/EtOAc) to afford methyl 4-bromo-2-nitrobenzoate as a pale yellow solid (3.52 g, 90%). To a solution of the nitrobenzoate (3.52 g, 13.5 mmol) in 1:1 EtOAc/DCM (30 mL) at rt was added SnCl2.2H2O (15.27 g, 67 mmol). After 18 h, the mixture was concentrated, diluted with satd. aq. NaHCO3, and extracted with DCM (3×). The combined organic layers were dried (MgSO4) and concentrated to provide the desired aminobenzoate as a white solid (2.89 g, 93%). 1H NMR (400 MHz, CDCl3): 7.70(d, J=8.6, 1H), 6.84 (d, J=1.9, 1H), 6.75 (dd, J=8.6, 1.9, 1H), 5.78 (br s, 2H), 3.86 (s, 3H). |
| 90 - 98% |
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In N,N-dimethyl-formamide; at 0 - 20℃; for 48.25 - 72.25h;Product distribution / selectivity; |
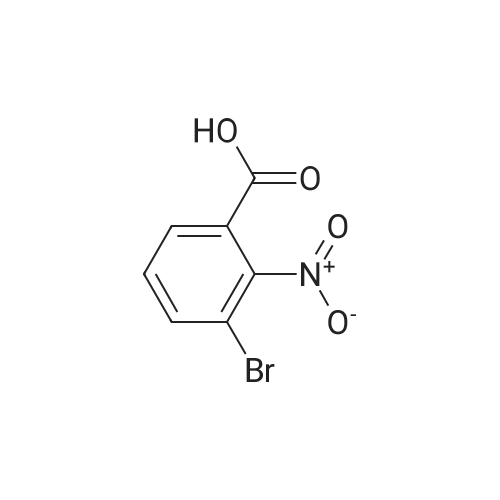
Example 62:; N-[7-ethyI-6-(l-methyI-lH-pyrazol-4-yl)-2,4-dioxo-l,4-dihydro-2H-quinazoIin-3-yI]- methanesulfonamide; 4-Bromo-2-nitro-benzoic acid methyl ester; To a solution of 4-bromo-2-nitrobenzoic acid (25.3 g, 103 mmol) in DMF (200 mL) at 0 0C were added DBU (79.1 mL, 514.8 mmol) and MeI (32.2 mL, 515 mmol). The reaction mixture was stirred for 15 min at this temperature and for 72 h at r.t. The mixture was poured into water and extracted with EtOAc (2X). The combined organic layers were washed with water (2X), dried (Na2SO4) and the solvent was removed in vacuo. The residue was purified by flash chromatography (silica gel, 7:3 hexanes:EtOAc) to provide the title compound (26.24 g, 98%) as a yellow oil.; Example 71:; N-[7-(l,l-difluoro-ethyl)-6-(2-isopropyl-2H-pyrazol-3-yl)-2,4-dioxo-l,4-dihydro-2H- quinazolin-3-yl]-methanesulfbnamide; 4-Bromo-2-nitro-benzoic acid methyl ester; To solution of 4-bromo-2-nitro-benzoic acid (9 g, 36.58 mmol) in dimethylforrnamide (36 mL) cooled to 0 0C were added l,8-diazabicyclo[5.4.0]und-7-ene (28.09 mL, 182.9 mmol) and MeI (11.4 mL, 182.5 mmol). The reaction mixture was stirred at 0 0C for 15 min and at r.t. for 48 h. The mixture was poured into water and extracted with EtOAc (2X). The combined organic phases were washed with water (2X), dried (Na2SO4) and concentrated to dryness. The crude product was purified by flash chromatography (hexanes to EtOAc / hexanes (4:6)) to give 4-bromo-2-nitro-benzoic acid methyl ester (8.62 g, 33.147 mmol, 90%). 1H-NMR (CDCl3, 400 MHz) 8.02 (s, 1 H), 7.81 (dd, J= 2.3, 10.5Hz IH), 7.66 (d, J= 8.2 Hz, IH), 3.92 (s, 3H). |
| 78% |
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 3h; |
To a solution of 4-bromo-2-nitrobenzoic acid (10 g, 40.6 mmol) in DMF (100 mL) was added potassium carbonate (11.24 g, 81 mmol) followed by methyl iodide (3.30 mL, 52.8 mmol) dropwise. The mixture was stirred at 80 C. for 3 h. After cooling to room temperature, the mixture was filtered and residue was dissolved in ethyl acetate (2*200 mL). The organic layer was washed with water (200 mL) followed by brine (100 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude sample was purified by flash chromatography (5% Ethyl acetate: Pet ether; 80 g silica gel column) to afford 1A (off white solid, 8.2 g, 31.5 mmol, 78% yield). 1H NMR (400 MHz, DMSO-d6) delta 8.35 (d, J=2.0 Hz, 1H), 8.07 (dd, J=8.0, 1.6 Hz, 1H), 7.84 (d, J=8.0 Hz, 1H), 3.85 (s, 3H). |
| 0.90% |
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In DMF (N,N-dimethyl-formamide); at 0 - 20℃; |
To a stirred solution of 4-bromo-2-nitrobenzoic acid (3.8 g, 15 mmol) in DMF (30 mL) at 0 C. was added 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (10.0 mL, 75.0 mmol) followed by iodomethane (4.7 mL, 75 mmol). The reaction mixture was stirred 15 min at 0 C., then was allowed to warm to room temperature and was stirred overnight. The mixture was poured into H2O and extracted with EtOAc (2×). The combined organic extracts were washed with H2O (2×), dried (MgSO4), and concentrated in vacuo. The residue was purified by flash chromatography (hexanes/EtOAc) to afford methyl 4-bromo-2-nitrobenzoate as a pale yellow solid (3.52 g, 0.90%). To a solution of the nitrobenzoate (3.52 g, 13.5 mmol) in 1:1 EtOAc/DCM (30 mL) at room temperature was added SnCl2.2H2O (15 g, 67 mmol). The reaction mixture was allowed to stir overnight. The solvents were evaporated in vacuo, and the residue was partitioned between satd. aq. NaHCO3 and DCM. The layers were separated, and the aqueous layer was further extracted with DCM (2×). The combined organic layers were dried (MgSO4) and concentrated in vacuo to provide the pure aminobenzoate as a white solid (2.89 g, 93%). 1H NMR (400 MHz, CDCl3): 7.70 (d, J=8.6 Hz, 1H), 6.84 (d, J=1.9 Hz, 1H), 6.75 (dd, J=8.6, 1.9 Hz, 1H), 5.78 (br s, 2H), 3.86 (s, 3H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping