| 65.94% |
With caesium carbonate; In tetrahydrofuran; acetonitrile; at 20℃; for 2h; |
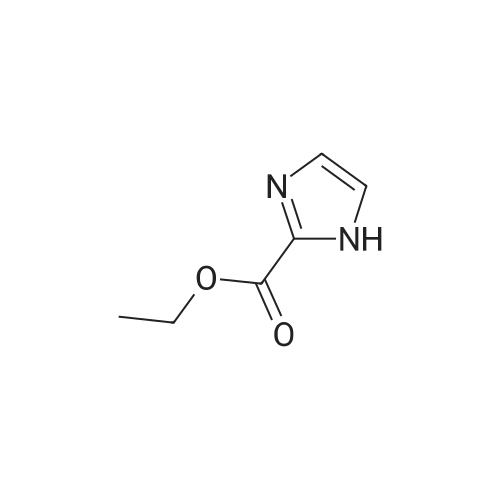
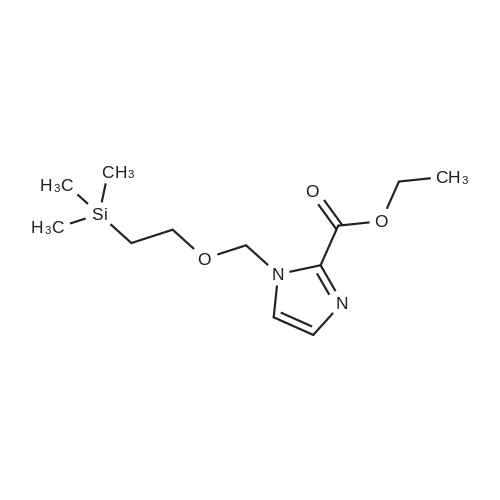
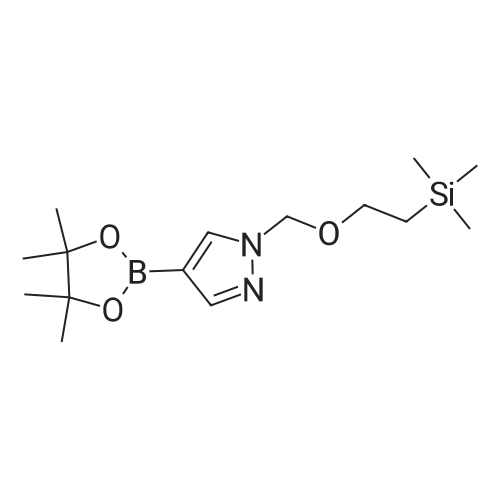
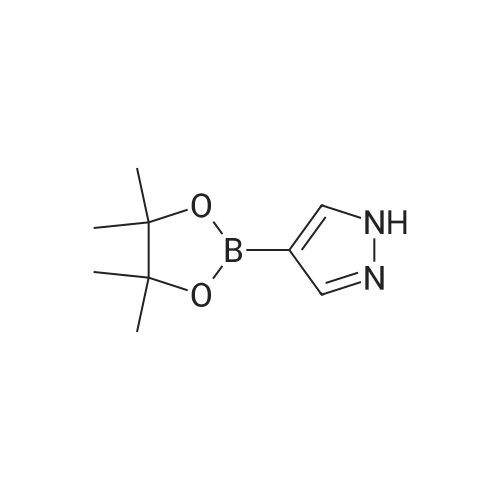
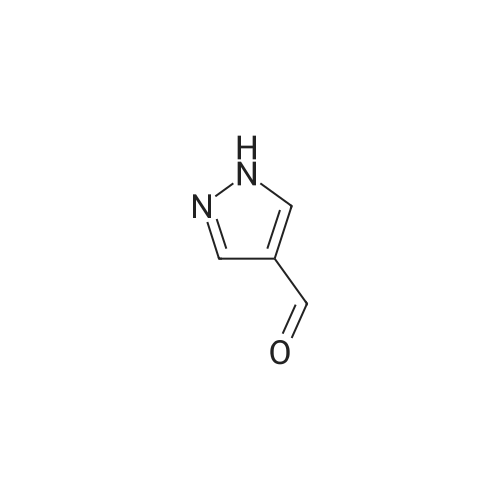
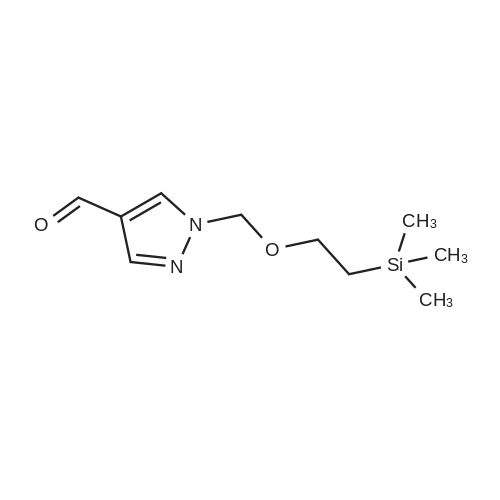
13.2 4-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)-1-(2-trimethylsilanyl- ethoxymethyl)-1 H-p razole To a solution of 1H-pyrazole-4-boronic acid pinacol ester (0.5 g, 2.57 mmol), in tetrahydrofuran/acetonitrile (3:2, 20ml), 2-(chloromethoxylethyl)trimethyl- silane (0.51 g, 3.09 mmol) and cesium carbonate (1.67 g, 5.15 mmol) are added and stirred for 2 hours at room temperature. The reaction mixture is filtered through celite, and concentrated, the crude mass is taken in ethylacetate (30 ml), washed with water, brine solution, dried over anhydrous MgS04 and concentrated to get the product as brown oil (0.55 g, 65.94 %); TLC: Pet ether/ethyl acetate(8/2) R - 0.5; 1H NMR: 400 MHz, DMSO-d6: delta [ppm] 8.08 (s, 1H), 7.64 (s, 1 H), 5.40 (s, 2H), 3.48-3.54 (m, 2H), 1.24 (s, 12H), 0.81-0.85 (m, 2H), -0.049(s, 9H); |
| 61% |
|
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (348 mg, 1.8 mmol) was dissolved in DMF (5 mL) and sodium hydride (60% dispersion, 86 mg, 2.15 mmol) added and the mixture heated to 60 C. for 5 min. Upon cooling and stirring for an additional 15 min, trimethylsilylethoxymethyl chloride (358 mg, 2.15 mmol, 381 muL) was added dropwise over 5 min and mixture stirred for 16 h. The reaction mixture was diluted with ethyl acetate (25 mL), washed with 5% lithium chloride (5×), dried over sodium sulfate and concentrated. The residue was purified by column chromatography (40 g ISCO column eluting with hexanes and ethyl acetate; gradient 100% hexanes to 50% hexanes over 30 min at 30 mL/min) to provide the SEM-protected pyrazole (360 mg, 61%) as a colorless oil; 1H NMR (500 MHz, CDCl3) delta 7.84 (s, 1H), 7.80 (s, 1H), 5.42 (s, 2H), 3.56-3.53 (t, J=8.3 Hz, 2H), 1.31 (s, 12H), 0.91-0.87 (t, J=8.3 Hz, 2H), -0.03 (s, 9H). |
| 56% |
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 3h; |
To a solution of methyl 4-(tetramethyl-1 ,3, 2-dioxaborolan-2-yl)-1H-pyrazole (1 .238 g, 6.316 mmol) in DMF (20 mL) was added potassium carbonate (2.62 g, 18.95 mmol) and [2-(chloromethoxy)ethyl](trimethyl)silane (1 .68 mL, 9.48 mmol) at room temperature. The mixture was stirred for 3 hours and then partitioned between TBME (100 ml.) and water (50 ml_). The organic layer was separated, washed with water (2 x 30 mL) and brine (30 ml_), dried (Na2S04) and concentrated at reduced pressure. The residue was purified by Biotage Isolera chromatography [Biotage SNAP Cartridge KP-Sil 50 g; using a gradient of eluents, 0-50% EtOAc in heptane]. The product containing fractions were combined, concentrated in vacuo to give the title compound (1 .20 g, 56% yield) as colourless oil. 1H NMR (500 MHz, chloroform-d) delta [ppm] 7.88 (s, 1 H), 7.84 (s, 1 H), 5.46 (s, 2H), 3.61- 3.55 (m, 2H), 1 .35 (s, 12H), 0.96 - 0.90 (m, 2H), 0.00 (s, 9H). LCMS (Analytical Method A): Rt = 1 .34 mins; MS (ESIPos) m/z = 324.95 (M+H) |
| 46% |
|
32-(a) 4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-1-(2-trimethylsilylethoxymethyl)-1H-pyrazole; Under argon atmosphere, to 20 ml of tetrahydrofuran solution containing 1.09 g (5.62 mmol) of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole was added 443 mg (11.1 mmol) of 60% sodium hydride under ice-cooling, and the mixture was stirred for 5 minutes. Then, 3 ml (17.0 mmol) of (2-trimethylsilylethoxy)methyl chloride was added dropwise to the mixture, and the mixture was reacted at room temperature for 2 hours. After completion of the reaction, water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was separated, and the solutions were washed successively with water and then with a saturated aqueous solution of sodium chloride, dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The obtained residue was applied to silica gel column chromatography (Eluent; hexane:ethyl acetate=9:1 (V/V)), and the fractions containing the desired compound were concentrated under reduced pressure to obtain 832 mg of the title compound as a colorless oil. (46%) Mass Spectrum (CI, m/z): 325 (M++1). 1H-NMR Spectrum (CDCl3, delta ppm): -0.03 (s, 9H), 0.86-0.94 (m, 2H), 1.32 (s, 12H), 3.51-3.59 (m, 2H), 5.43 (s, 2H), 7.81 (d, J=0.5 Hz, 1H), 7.86 (d, J=0.5 Hz, 1H). |
| 44% |
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃; for 3h; |
To a stirred solution of 4-(4 , 4,5 , 5-tetra methyl- 1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (2 g, 10.3 mmol) in DMF (30 mL), (2-(chloromethoxy)ethyl)trimethylsilane (2 g, 12.3 mmol), and CS2CO3 (10 g, 30.9 mmol) were added. The resulting mixturen was stirred at rt for 3 h. Solvents were evaporated and the crude residue was diluted with ice cold water and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SC>4 and filtered. The filtered solution was concentrated under reduced pressure and the resulting crude compound was purified by flash column chromatography using 20-30% EtOAc/Pet ether to get the title compound (1.5 g, 44%) as pale yellow gummy.LC-MS (method 14): R, = 3.08 min; m/z = 325.2 (M+H?). |
|
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; at 25℃; for 0.75h; |
Preparative Example 1; To a suspension of potassium carbonate (5.85 g, 1.5 equiv) and 4- (4,4,5,5-tetramethyM ,3,2-dioxaborolan-2-yl)-1 h-pyrazole (5.48 g, 1.0 equiv) in NMP (50 mL) at rt was added SEMCI (5.2 mL, 1.05 equiv) dropwise (mildly exothermic). The resulting mixture was allowed to stir an additional 45 min at rt. The reaction was diluted with ethyl acetate, rinsed with water (2x), brine and dried (sodium sulfate). Filtration and concentration afforded the title compound that was used without purification. MH+ = 325. |
|
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; for 1h; |
A mixture of 4,4,5,5-tetramethyl-2-(1H-pyrazol-4-yl)-1,3,2-dioxaborolane (5.48 g), SEMCl (5.2 mL), and K2CO3 (5.85 g) in NMP (50 mL) was stirred under N2 for 1 hr. The reaction mixture was diluted with EtOAc, rinsed with H2O, brine, and dried over Na2SO4. The mixture was filtered, the solvents were evaporated and the residue was used directly in the next step. |
|
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; at 20℃; for 0.75h; |
To a suspension of potassium carbonate (5.85 g, 1.5 equiv) and 1H-pyrazole-4-boronate (5.48 g, 1.0 equiv) in NMP (50 mL) at room temperature was added SEMCI (5.2 mL, 1.05 equiv) dropwise (mildly exothermic). The resulting mixture was allowed to stir for an additional 45 min at room temperature. The reaction was diluted with ethyl acetate, rinsed with water (×2), brine and dried (sodium sulfate). Filtration and concentration afforded the title compound (270) that used directly in the next step. |
|
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; at 20℃; for 0.75h; |
Example 270; To a suspension of potassium carbonate (5.85 g, 1.5 equiv) and 1H-pyrazole-4-boronate (5.48 g, 1.0 equiv) in NMP (50 mL) at room temperature was added SEMCl (5.2 mL, 1.05 equiv) dropwise (mildly exothermic). The resulting mixture was allowed to stir for an additional 45 min at room temperature. The reaction was diluted with ethyl acetate, rinsed with water (×2), brine and dried (sodium sulfate). Filtration and concentration afforded the title compound (270) that used directly in the next step. |
|
|
Preparation E4-(4A5 -tetramethyl-l ,3,2-dioxaborolan-2-yl)-l-((2-(trimethylsilyl)ethoxy)methyl)-lH- pyrazole [00624] A solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole(10.00 g, 51.54 mmol) in DMF (100 mL) was cooled to 0 C in an ice bath and treated with sodium hydride (60% dispersion in oil) (3.092 g, 77.30 mmol) in one portion. The reaction mixture was stirred at 0 C for 2 minutes, then at ambient temperature for 30 minutes. The reaction mixture was cooled to 0 C and (2-(chloromethoxy)ethyl)trimethylsilane (11.82 mL, 67.00 mmol) was added. The reaction mixture was warmed to ambient temperature and allowed to stir overnight. The reaction mixture was poured into aqueous saturated ammonium chloride (100 mL) containing ice (approximately 100 mL) and stirred until the ice melted. After the ice melted, the cold mixture was extracted with ethyl acetate. The combined organic extracts were washed with water, brine, dried over MgS04, and concentrated under reduced pressure to afford the title compound (14.45 g, 44.56 mmol, 86.46% yield). MS (apci) m/z = 325.0 (M+H). |
|
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; at 20℃; for 16h;Inert atmosphere; |
To a solution of 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-lH-pyrazole (8.2 g, 41 mmol) in NMP (60 mL) were added K2C03 (12 g, 82 mmol) and 2-(trimethylsilyl)ethoxymethyl chloride (7.8 mL, 43 mmol) in sequence. The reaction mixture was stirred at RT under N2 for 16 h. Then, the reaction mixture was diluted filtered and the filtrate was diluted with EtOAc (300 mL), The resulting solution was washed with sat NaHC03(aq) (3 x 200 mL), ?0 (4 x 200 mL), brine (1 x 200 mL), dried over Na2S0 , filtered, concentrated and dried in vacuo to yield Int-37 as clear yellowish oil |
| 0.9 g |
|
A) 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole [1064] To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (770 mg) in DMF (10 mL) was added sodium hydride (60%, 114 mg) under ice-cooling. The reaction mixture was stirred at room temperature for 1 hr. To the reaction mixture was added dropwise (2-(chloromethoxy)ethyl)(trimethyl)silane (990 mg) at room temperature, and the mixture was stirred for 15 hr. The reaction mixture was diluted with ethyl acetate, and the mixture was washed with 5% aqueous lithium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give the title compound (0.90 g). MS(ESI+): [M+H]+ 325.2. MS(ESI+), found: 325.2. |
|
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 16h; |
To a solution of 4-(4,4,5,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole (1.00 g, 5.15 mmol) in DMF (20 mL) was added K2C03 (1.07 g, 7.73 mmol) and (2-(chloromethoxy)ethyl)trimethylsilane (1.03 g, 6.18 mmol). The reaction was stirred at ambient temperature for 16 h and then diluted with water (20 mL). The mixture was extracted with EtOAc (2 x 30 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the crude 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy) methyl)-1H-pyrazole (1.00 g, 60% yield) as ayellow oil.A mixture of 2-(3-bromophenyl)- 1,1,1 -trifluorobut-3 -yn-2-ol (500 mg, 1.79 mmol), 4-(4,4,5,5- tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 -((2-(trimethylsilyl)ethoxy)methyl)- 1 H-pyrazole (872 mg, 2.69 mmol), Cs2CO3 (1.17 g, 3.58 mmol) and 1,1?-Bis(diphenylphosphion) ferrocene dichloride palladium(II) (66 mg, 0.09 mmol) in dioxane/H20 (5 mL/1 mL) was heated at 110 Cfor 0.5 h under microwave conditions. The solvent was removed and the crude residue was purified by column chromatography (petroleum ether: EtOAc = 4:1) to give the title compound (300 mg, 42% yield) as a colorless oil. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping