Abstract: In our previous study, riluzole azo-linked to salicylic acid (RAS) was prepared as a colon-targeted prodrug of riluzole (RLZ) to facilitate the repositioning of RLZ as an anticolitic drug. RAS is more effective against rat colitis than RLZ and sulfasalazine, currently used as an anti-inflammatory bowel disease drug. The aim of this study is to further improve colon specificity, anticolitic potency, and safety of RAS. N-succinylaspart-1-ylRLZ (SAR) and N-succinylglutam-1-ylRLZ (SGR) were synthesized and evaluated as a ""me-better"" colon-targeted prodrug of RLZ against rat colitis. SAR but not SGR was converted to RLZ in the cecal contents, whereas both conjugates remained intact in the small intestine. When comparing the colon specificity of SAR with that of RAS, the distribution coefficient and cell permeability of SAR were lower than those of RAS. In parallel, oral SAR delivered a greater amount of RLZ to the cecum of rats than oral RAS. In a DNBS-induced rat model of colitis, oral SAR mitigated colonic damage and inflammation and was more potent than oral RAS. Moreover, upon oral administration, SAR had a greater ability to limit the systemic absorption of RLZ than RAS, indicating a reduced risk of systemic side effects of SAR. Taken together, SAR may be a "me-better" colon-targeted prodrug of RLZ to improve the safety and anticolitic potency of RAS, an azo-type colon-targeted prodrug of RLZ.
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
RDCCTOOKKCKDKQCKOQRCCA-NH2, containing three disulfide bonds between Cys<SUP>3</SUP> and Cys<SUP>15</SUP>, Cys<SUP>4</SUP> and Cys<SUP>20</SUP>, Cys<SUP>10</SUP> and Cys<SUP>21</SUP>, O: 4-trans-hydroxyproline<SUP> </SUP>[ No CAS ]
RDCCTOOKKCKDRQCKOQKCCA-NH2, containing three disulfide bonds between Cys<SUP>3</SUP> and Cys<SUP>15</SUP>, Cys<SUP>4</SUP> and Cys<SUP>20</SUP>, Cys<SUP>10</SUP> and Cys<SUP>21</SUP>, O: 4-trans-hydroxyproline[ No CAS ]
KDCCTOOKKCKDKQCKOQRCCA-NH2, containing three disulfide bonds between Cys<SUP>3</SUP> and Cys<SUP>15</SUP>, Cys<SUP>4</SUP> and Cys<SUP>20</SUP>, Cys<SUP>10</SUP> and Cys<SUP>21</SUP>, O: 4-trans-hydroxyproline[ No CAS ]
KDCCTOOKKCKDRQCKOQKCCA-NH2, containing three disulfide bonds between Cys<SUP>3</SUP> and Cys<SUP>15</SUP>, Cys<SUP>4</SUP> and Cys<SUP>20</SUP>, Cys<SUP>10</SUP> and Cys<SUP>21</SUP>, O: 4-trans-hydroxyproline[ No CAS ]
5,8,11,14,17,20,23,26-octaoxa-2-azanonacosanedioic acid, 1-(9H-fluoren-9-ylmethyl) ester[ No CAS ]
[ 4530-20-5 ]
[ 2488-15-5 ]
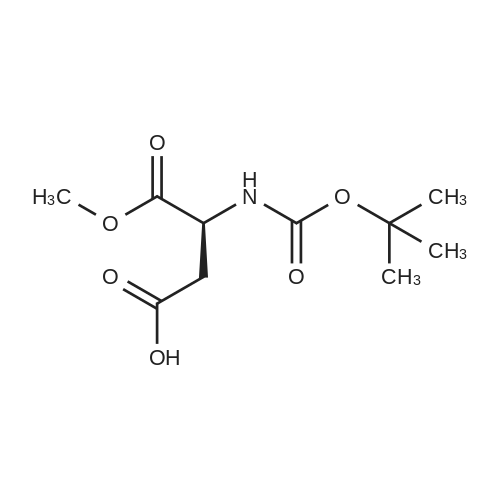
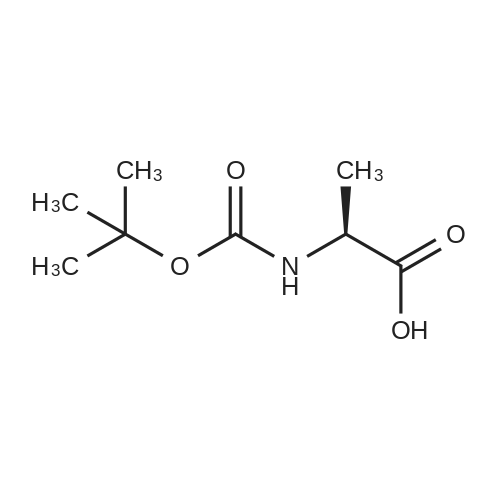
[ 7536-58-5 ]
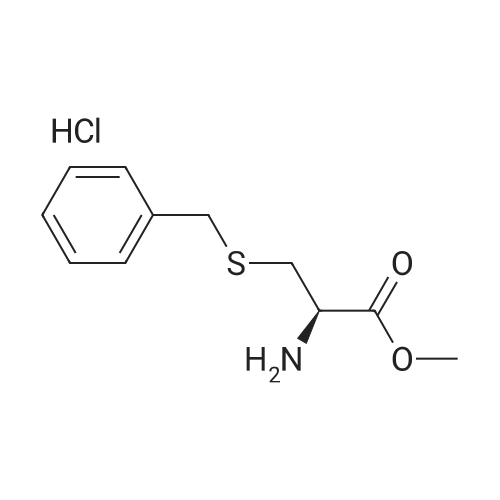
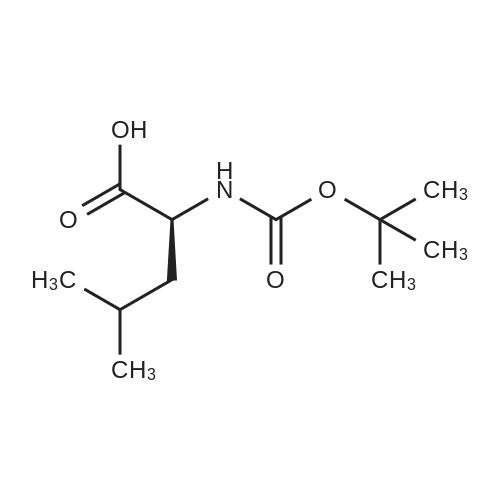
[ 13734-34-4 ]
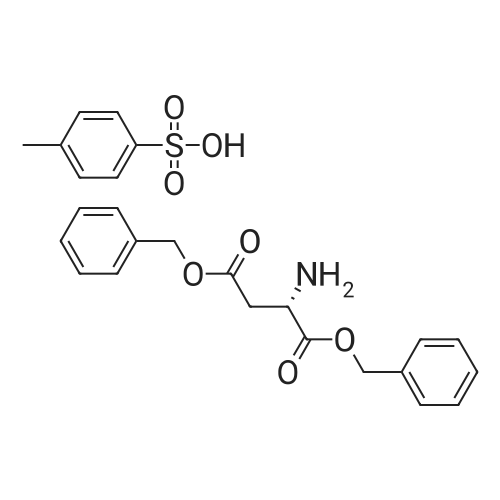
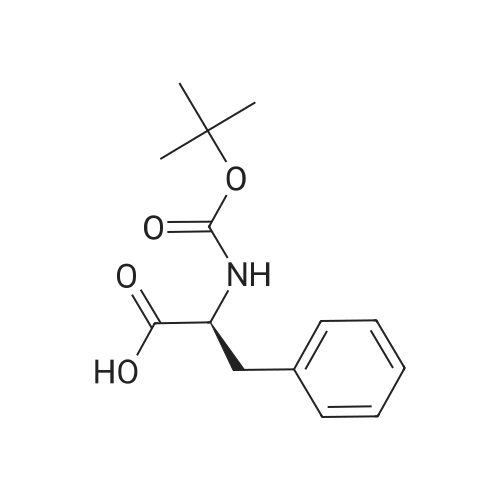
[ 47355-10-2 ]
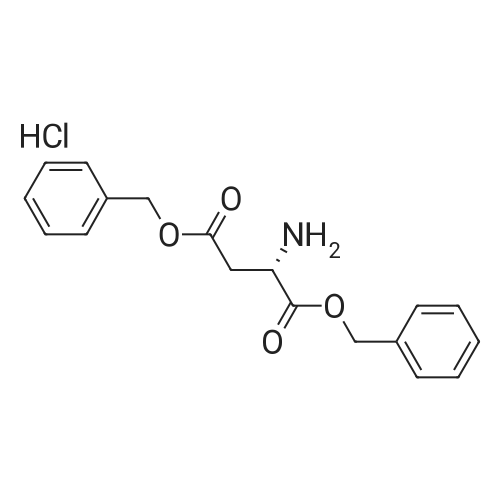
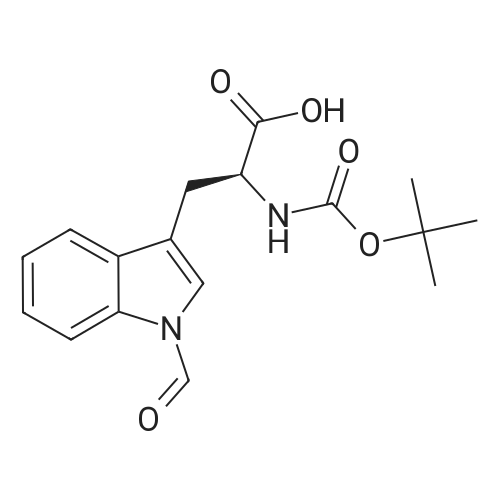
[ 57-10-3 ]
C70H111N9O20S[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: The second portion of the CCK4 peptide and the SubP peptide were modified on resin as follows to yield test lipidated peptides (1-SubP-COOH and 1-CCK4-Gly-COOH). Spacers (these are AA's used between the peg linker and the peptide of interest) were introduced on the peptides before pegylation (KGG for SubP and GG for CCK4). The free N-terminus of the peptide on resin was first pegylated with N-Fmoc-PEG8-propionic acid using standard HBTU coupling conditions. The N-Fmoc protecting group was removed by treatment with 10% piperidine in DMF (N,N-Dimethylformamide) for 5 min. Palmitic acid was subsequently coupled with the N-terminal free amine of the pegylated peptide. Peptides were cleaved from the resin using high HF conditions with minor modifications to the usual procedure. For the SubP peptide, longer times were used to ensure removal of Arg(Tos) protecting group (90% anhydrous HF/10% anisole at 0 C. for 2 h). For the CCK-4 peptides, 1,3-propanedithiol was used in the HF cleavage mixture to ensure deprotection of the formyl protecting group and prevent oxidation of methionine to its sulfoxide derivative: 85% anhydrous HF/10% anisole/5% PDT (1,3-propaneithiol) at 0 C. for 2 h) (Matsueda, 1982). Following cleavage from resin, peptides were precipitated with cold Et2O. Unmodified peptides were extracted using 10% AcOH in water and the lipidated peptides were extracted using 10% AcOH in H2O followed by 10% AcOH in 50% EtOH/H2O. Crude peptides were purified by RP-HPLC [Vydac C18, 10 m, 22 mm×250 mm]. The purities of the peptides were assessed by analytical RP-HPLC [Vydac C18, 5 m, 4 mm×250 mm].
Peptides illustrated in FIG. 9 were synthesized using the in-situ neutralization protocol for t-Boc chemistry (Schnolzer et al., In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences, 1992, International journal of peptide and protein research 40:180-193) on PAM resin on a 0.5 mmol scale. Amino acids were used with the following side chain protecting groups: Arg(Tos), Asp(OBzl), Gln(Xan), Lys(Fmoc), Lys(2-Cl-Z) and Trp(For). Peptide coupling reactions were carried out with a 4-fold excess (2.0 mmol) of activated amino acid for at least 15 min. The t-Boc protecting group on the N-terminus was removed using trifluoroacetic acid (TFA). The PAM resin from the CCK4 peptide synthesis was split into two equal portions. One portion of the resin was used for synthesizing non-lipidated peptides. The CCK4 (s-CCK-Gly-COOH) peptide was left unmodified at the N-terminus. This peptide served as positive control for the lipidated counterparts.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping