|
|
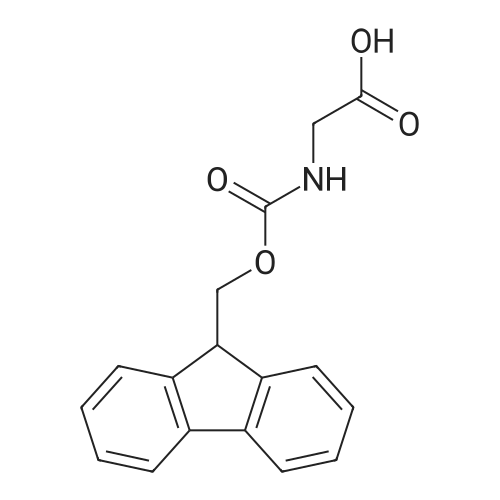
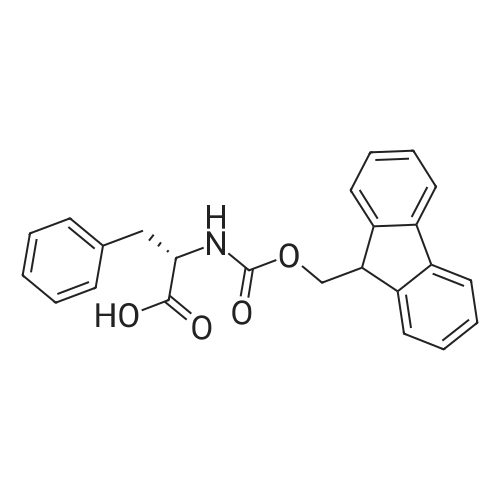
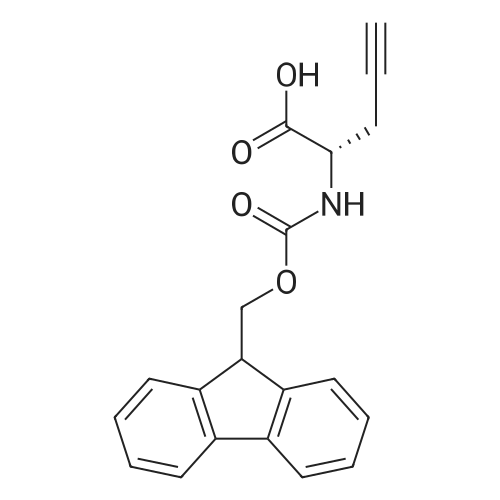
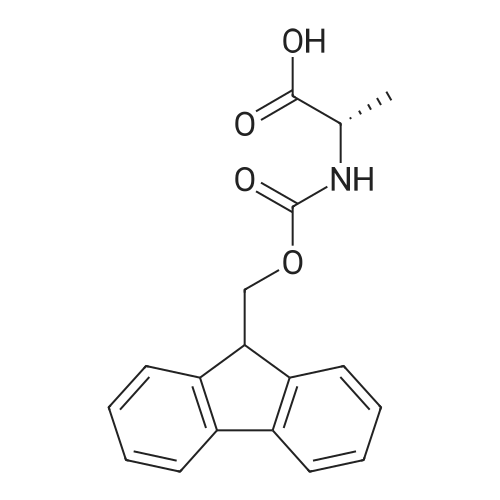
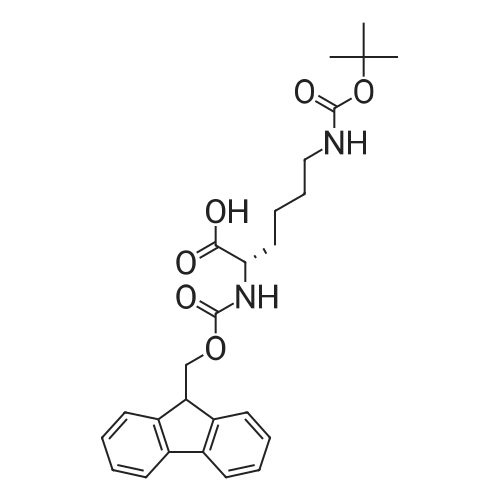
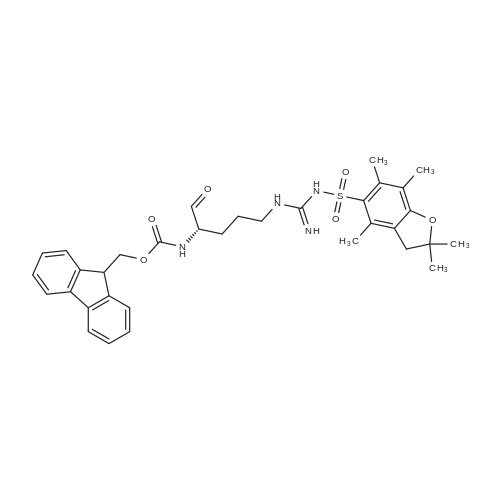
The solid phase peptide synthesis (SPPS) was performed using a microwave assisted protocol (Discover microwave oven, CEMCorp.) starting from Fmoc-Leu-Wang resin. The reactions were carried out in a silanized glass tube loosely sealed with a silicon septum. Remark: the development of overpressure was avoided by using DMF as the solvent and intermittent cooling in an ethanol-ice bath. The amino acids were incorporated as their commercially available derivatives in the following order: Fmoc-Ile-OH, Fmoc-N-homo-Tyr(tBu)-OH (synthesized according to ref. 18), Fmoc-Pro-OH, Fmoc-Arg(Pbf)-OH, Fmoc-N-Me-Arg(Mtr)-OH and Fmoc-propargyl-Gly-OH. Elongation of the peptide chain was performedby repetitive cycles of Fmoc deprotection and subsequent couplings of the amino acid. Fmoc deprotection was performed by treating the resin with 25% piperidine in DMF (microwave irradiation: 7 5 s, 100 W), followed by washings with DMF (5). In between each irradiation step, cooling of the reaction mixture to a temperature of 10 C was achieved by sufficient agitation in an ethanol-ice bath. Peptide couplings of Fmoc-Ile-OH, Fmoc-Arg(Pbf)-OH and Fmoc-N-Me-Arg(Mtr)-OH were performed employing 5 equiv of each Fmoc-AA/PyBOP/DIPEA and 7.5 equiv 1-hydroxybenzotriazole (HOBt), dissolved in a minimum amount of DMF (irradiation: 20 10 s, 50W and intermittent cooling). Fmoc-N-homo-Tyr(tBu)-OH (3 equiv) was coupled with 3 equiv PyBOP/DIPEA and 4.5 equiv HOBt in DMF. Fmoc-Pro-OH (5 equiv)and Fmoc-propargyl-Gly-OH (5 equiv) were subjected to a double coupling with HATU (5 equiv) and DIPEA (10 equiv) in DMF. After the last acylation step, the N-terminal Fmoc-residue was deprotected, the resin was 10 rinsed with CH2Cl2 and dried in vacuo. The cleavage from the resin was performed using a mixture of trifluoroacetic acid (TFA)/phenol/H2O/triisopropylsilane (TIS) 88:6:4:2 for 4 h, followed by a filtration of the resin. After evaporation of the solvent in vacuo and precipitation in t-butylmethylether, the crude peptides were purified using preparative RP-HPLC (Agilent 1100 preparative series, column Zorbax Eclipse XDB-C8, 21.2 mm, 150 mm, 5 lm particles, flow rate 10 mL/min) with the solvent system 3-35% acetonitrile in water (0.1% HCO2H) in a linear gradient over 18.0 min, tR: 10.5 min. After the separation, the peptide was lyophilized and peptide purity and identity were assessed by analytical HPLC (Agilent 1100 analytical series, equipped with QuatPump and VWD detector; column ZorbaxEclipse XDB-C8 analytical column, 4.6 mm, 150 mm, 5 lm, flow rate 0.5 mL/min) coupled to a Bruker Esquire 2000 mass detector equipped with an ESI-trap. ESI-TOF high mass accuracy and resolution experiments were performed on a BRUKER maXis MS (BrukerDaltonics, Bremen) in the laboratories of the Chair of OrganicChemistry (Prof. Dr. Rik Tykwinski), Department of Pharmacy and Chemistry, Friedrich-Alexander University of Erlangen Nuremberg. Purity: solvent system 1: 10-55% methanol in water (0.1% HCO2H)in a linear gradient over 18 min, tR = 14.6 min (>99 %); solvent system 2: 3-40% acetonitrile in water (0.1% HCO2H) in a linear gradient over 26 min, tR = 15.9 min (>99%). ESI-MS: m/z calcd:940.6, found: 940.6 [M+H]+; HR-ESI-TOF: [M+H]+ calcd forC45H74N13O9: 940.5732, found: 940.5724. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping