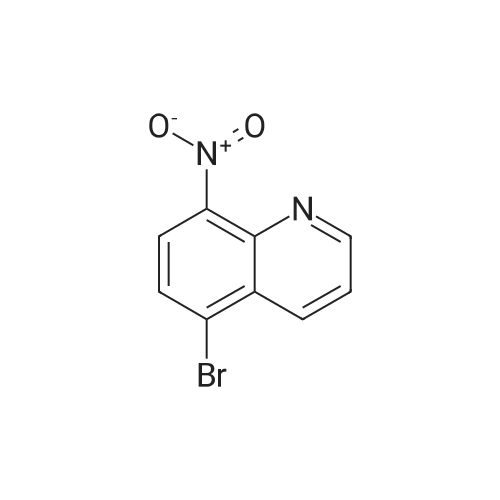
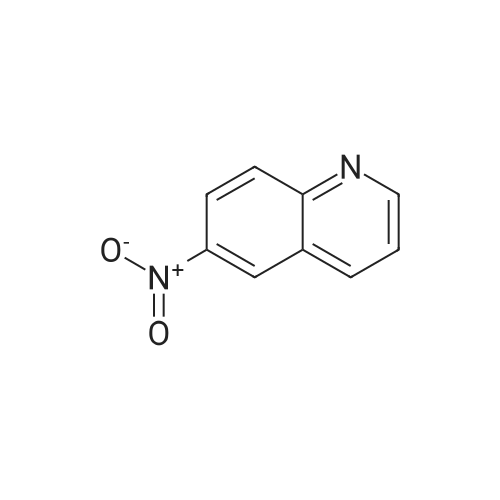
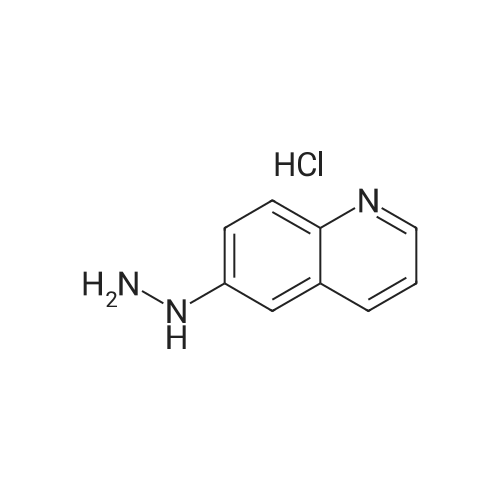
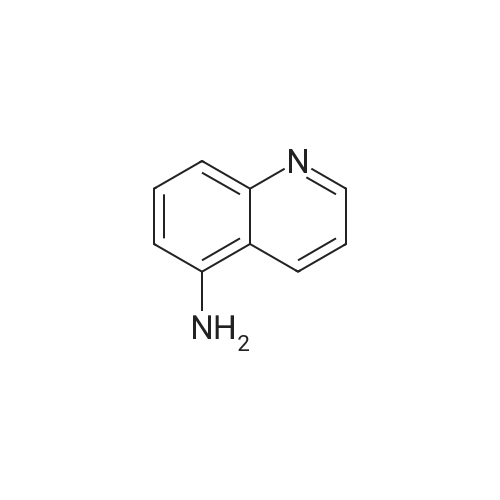
| 93% |
With palladium diacetate; hydrazine hydrate monohydrate; potassium hydroxide; In lithium hydroxide monohydrate; at 50℃; for 8h;Inert atmosphere; Green chemistry; |
General procedure: To an oven-dried reaction flask with Teflon coated stir bar purgedwith argon, Pd(OAc)2 0.01 mmol, IL4 0.08 mmol, KOH (or K2CO3)0.30 mmol, nitroarene 1 1.0 mmol and degassed HPLC grade water1.5 mL were added, and the mixture was stirred for 5 min under argonat room temperature. Then, 0.5 mL of hydrazine hydrate aqueous solution(~5.0 mmol) was syringed into the flask at the same temperature.After stirring for additional 5 min, the reaction was heat to 50 C,and stirring for 8 h under argon. After completion, the reaction mixturewas extracted by methyl tertiary butyl ether (MTBE) (3 × 2 mL), andthe organic layer was collected and filtered through a bed of silica gellayered over Celite. The volatiles were removed in vacuo to afford theproduct 2. In some cases, further column chromatography on silica gelwas required to afford the pure desired products. |
| 91% |
With hydrogen; In methanol; at 20℃; |
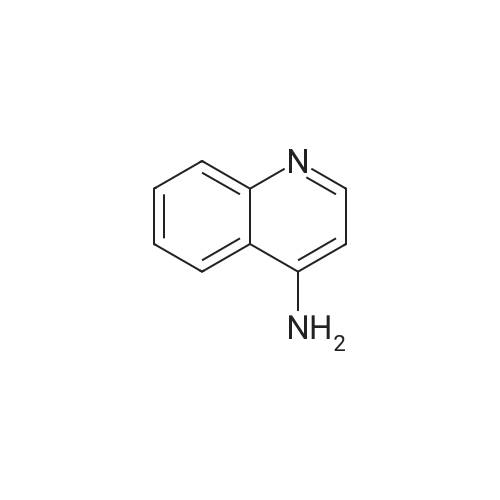
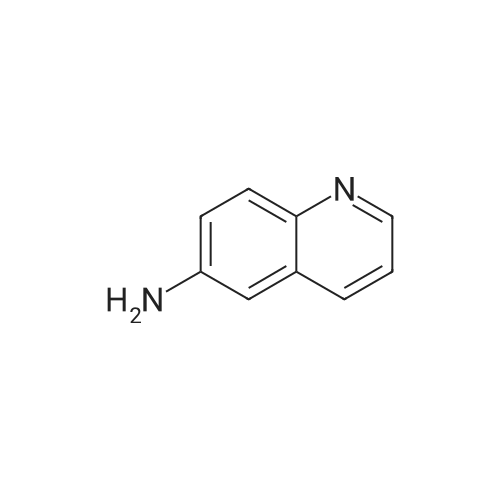
The solution of 6-nitroquinoline (174.2 mg, 1 mmol) in methanol (10 mL) wastreated with Pd/C (10%, 14.0 mg). the reaction mixture wasvigorously stirred under an atmosphere of hydrogen at roomtemperature. +e progress of the reaction was monitored byTLC. After completion of the reaction, the reaction mixturewas then filtered and concentrated in vacuo to give the crudeproduct. The crude product was purified by column (ethylacetate : petroleum ether: 4) to afford the product 2(131.1 mg, 91.0%) as white solid. m.p. 116.8-119.0C. 1HNMR (600 MHz, CDCl3) 2: 8.64 (d, J 4.0 Hz, 1H), 7.89 (t,J 9.7 Hz, 2H), 7.25 (t, J 4.8 Hz, 1H), 7.14 (dd, J 9.0 Hz,2.6 Hz, 1H), 6.88 (d, J 2.5 Hz, 1H), 3.84 (brs, 2H). |
| 90% |
With tetrahydroxydiborane; copper (II) acetate; In acetonitrile; at 100℃; for 24h;Inert atmosphere; |
6-nitroquinoline (0.6mmol, 114.1mg), Cu(OAc)2 (0.03mmol, 6.0mg) and tetrahydroxydiboron (0.9mmol, 80.7mg), acetonitrile (1mL), under nitrogen protection, 100 C After reacting for 24 hours, the reaction was monitored by TLC, 10 mL of water was added, ethyl acetate (10 mL×3) was added, the organic phases were combined, dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and separated by column chromatography (V petroleum ether: V ethyl acetate = 3:1) to obtain 77.9 mg of white solid, which is to obtain the target compound with a yield of 90%. |
| 87% |
With hydrazine hydrate monohydrate; In ethanol; at 80℃; for 1h;Inert atmosphere; |
General procedure: Hydrazine hydrate was chosen as the hydrogen donor for the low emission of pollutants. In a typical procedure, hydrazine hydrate (4 equiv) was added into the reactor which containing fresh prepared catalyst as described above. Then the reactor was put into a preheated oil bath with a stirring speed of 500 rpm, and the substrate (1 mmol)dissolved in 1 mL ethanol was added drop-wisely under argon. The reactions were monitored by TLC. After the reaction, the reaction mixture was vacuum filtered through a pad of silica on a glass-fritted funnel and an additional 15 mL of ethyl acetate (5 mL portions) was used to rinse the product from the silica, the filtrate was concentrated in vacuum and analyzed by GC. Products were purified by column chromatography and identified by 1H NMR and 13C NMR. |
| 83% |
With tetrahydroxydiborane; 5%-palladium/activated carbon; lithium hydroxide monohydrate; In acetonitrile; at 50℃; for 24h; |
General procedure: Nitrobenzene (0.6mmol), 5wt% Pd/C (0.5mmol %, 0.003mmol), H2O (10 equiv, 6.0mmol), B2(OH)4 (3.3 equiv, 2.0mmol), and CH3CN (1.0mL) were added in a 10mL tube. The reaction mixture was stirred at 50C for 24h. When the reaction was complete monitored by TLC, the mixture was cooled to room temperature. Water (5mL) was added, and extracted with EtOAc (3×5mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give aniline 2a (55mg, 99%). |
| 83% |
|
General procedure: The nitro compound (1 equiv), HFIP (10 equiv), Fe powder (5 equiv) were mixed in a tube. Then 2 N HCl aqueous solutions was added to the reaction mixture. After stirring at room temperature for 30 min, the reaction mixture was neutralized with sat. NaHCO3 (aq.) and extracted with EtOAc three times. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was then purified by column chromatography on silica gel to furnish the desired amine product. |
| 75% |
With tetrahydroxydiborane; copper (II) acetate; In acetonitrile; at 80℃; for 24h;Schlenk technique; |
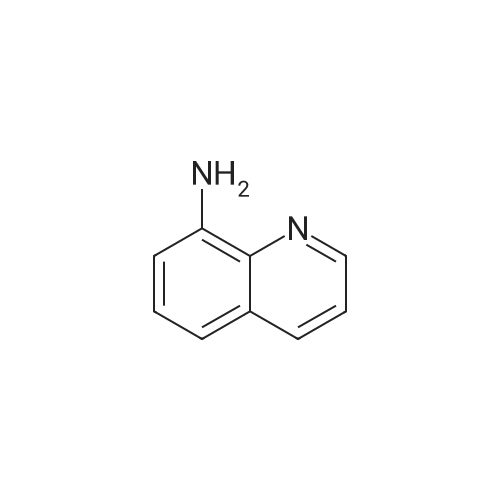
General procedure: A 20 mL Schlenk tube was charged with 8-nitroquinoline (1k; 87 mg, 0.5 mmol), Cu(OAc)2 (4.5 mg, 0.025 mmol), B2(OH)4(135 mg, 1.5 mmol), and MeCN (2.0 mL). The mixture was stirred at 80 C for 24 h, then cooled to room temperature and concentrated under reduced pressure. Similar workup to 2a gave a brown solid (4a: 63 mg, 87% yield). |
| 65% |
With tetrahydroxydiborane; lithium hydroxide monohydrate; at 80℃; for 8h; |
General procedure: Nitro aromatic (1.0 mmol), B2(OH)4 (5.0 equiv, 5.0 mmol), and H2O (3.0 mL) were added in a10 mL tube. The reaction mixture was stirred at 80 C for 8 h. When the reaction was completemonitored by TLC, the mixture was cooled to room temperature, extracted with ethyl acetate (3 ×20 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, andconcentrated under reduced pressure. The residue was purified by silica gel columnchromatography. |
| 46% |
With hydrogen;palladium 10% on activated carbon; In methanol; at 20℃; for 24h; |
INTERMEDIATE 1 Synthesis of 6-Amino-quinoline A suspension of 6-nitro-quinoline (8.7 g, 5 mmol), palladium on charcoal (10 %) (0.1 g) in methanol (0.2 L) was hydrogenated at room temperature for 24 with stirring. The catalyst was filtered and the solvent evaporated to yield a yellow solid. Crystallisation from ethyl acetate yielded the pure title compound as a pale yellow solid (3.3 g, 46 %). MS m/z: 145 [[M+H+]. IH] NMR (270 MHz, [CHC13-D)] [8] ppm 3.89 (s, 2 H) 6.87 (d, [J=2.] 64 Hz, 1 H) 7.14 [(DD,] [J=8.] 97,2. 64 Hz, 1 H) 7.25 [(DD,] [J=8.] 44,4. 22 Hz, [1] H) 7. [88 (DD, J=7.] 92,1. [58] Hz, 1 H) 7.90 (d, [J=8.] 97 Hz, 1 H) 8.63 [(DD,] [J=4.] 22,1. [58] Hz, 1 H). |
| 98%Chromat. |
With hydrazine hydrate monohydrate; In ethanol; at 70℃; for 1h;Schlenk technique; Sealed tube;Catalytic behavior; |
General procedure: Pd cNPs/CFe3O4 (20mg, 0.73mol% of Pd), aromatic nitro compounds (1mmol),N2H4.H2O (3mmol), and EtOH (3mL) were taken in a schlenk tube with a teflon stopcock, sealed and heated at 70C for a given time with constant stirring. After the completion of reaction, the catalyst was separated by an external magnet and reaction mixture was decanted. The solvent was evaporated and the residue was subjected to GC analysis (retention time of nitroaromatic compounds was used as internal standard) followed by column chromatography for further purification. The purified compounds were characterized by 1H NMR spectroscopy using CDCl3 as solvent and TMS as internal standard. The spectral details and spectra are given in supporting information section (Fig. S4a-S4l). |
|
With hydrogen; In methanol; at 120℃; under 11251.1 Torr; for 3h; |
General procedure: Typically, 0.5 mmol of nitrobenzene, 20 mg of Co-NSPC-X catalyst and 5 mL of methanol were added into a 30 mL stainless steel autoclave, and the autoclave was pressurized to 1.5 MPa. Then the reactor was placed into oil bath and heated to 120 C with vigorous stirring. After 2 h, the autoclave cooled down to room temperature and the hydrogen gas was carefully released. After the catalysts were removed by filtration, the remaining liquid mixture was analyzed on GC 9890 equipped with a DB-1701 column and a flame ionization detector (FID). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping