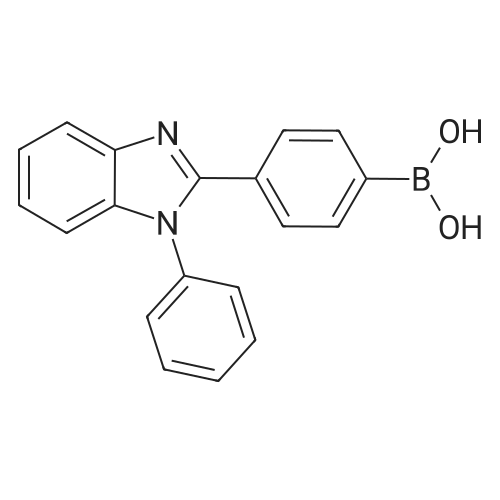
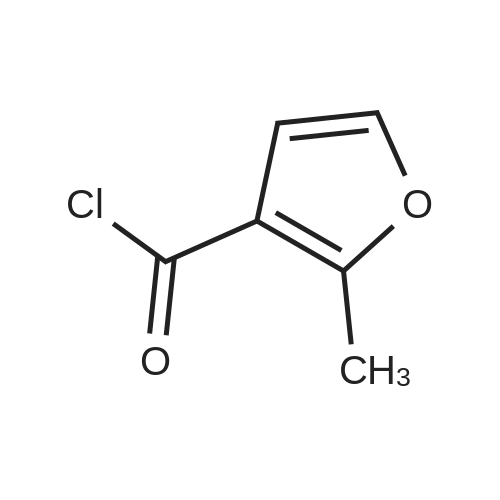
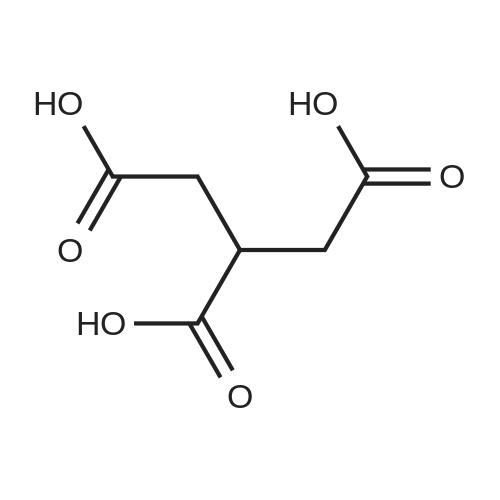
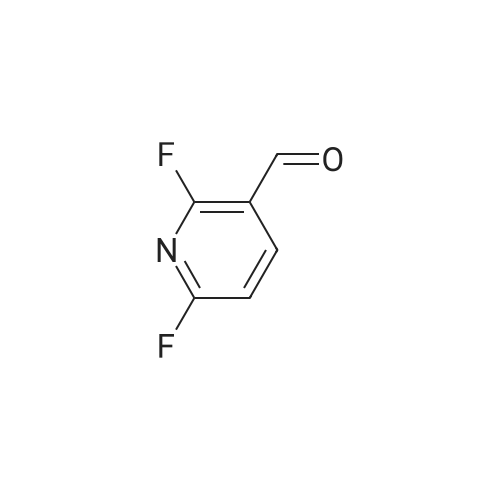
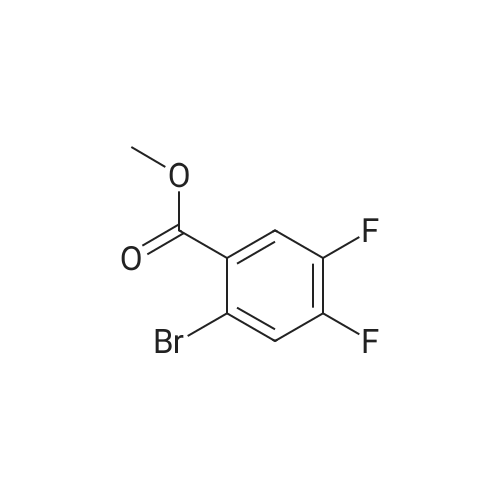
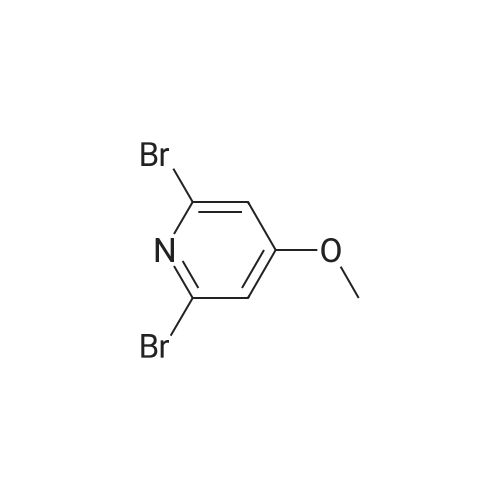
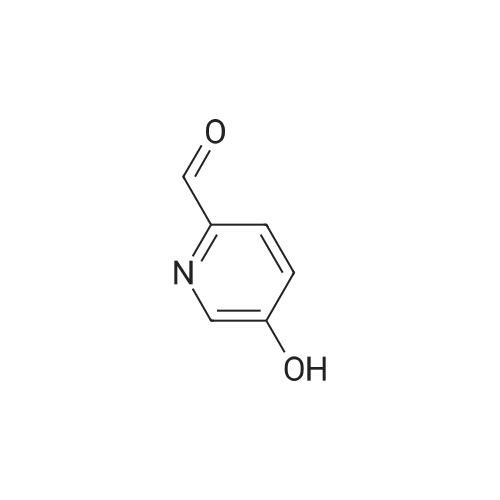
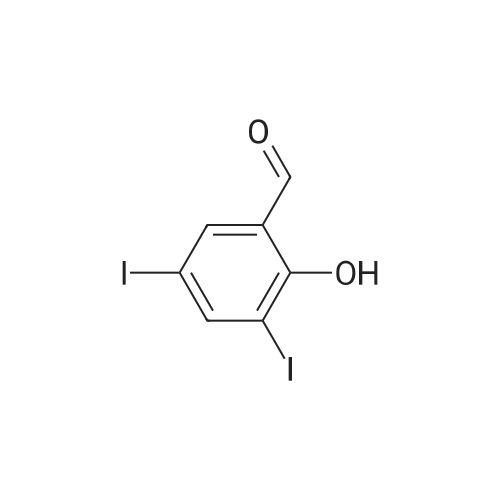
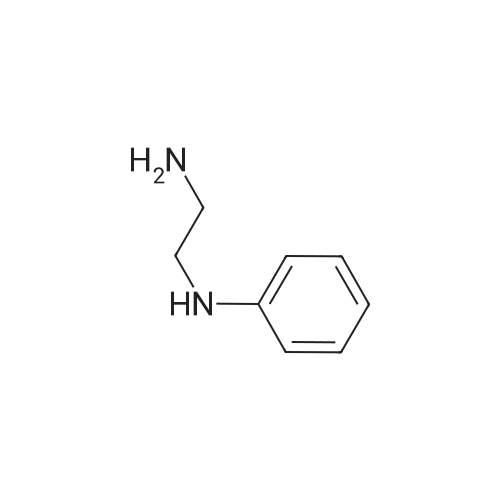
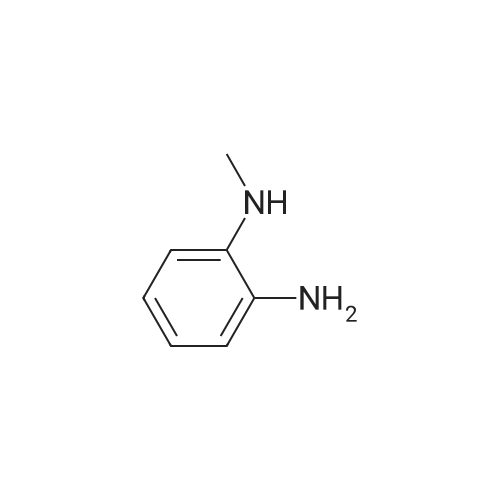
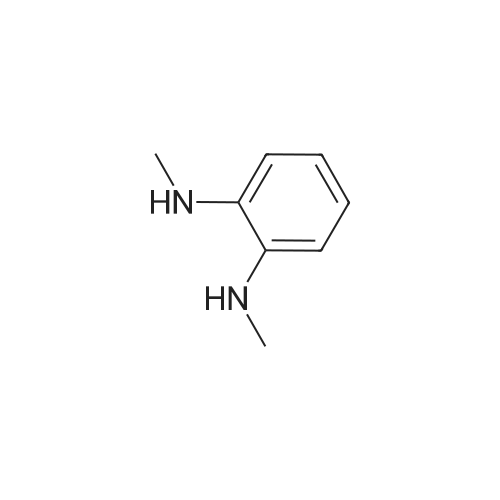
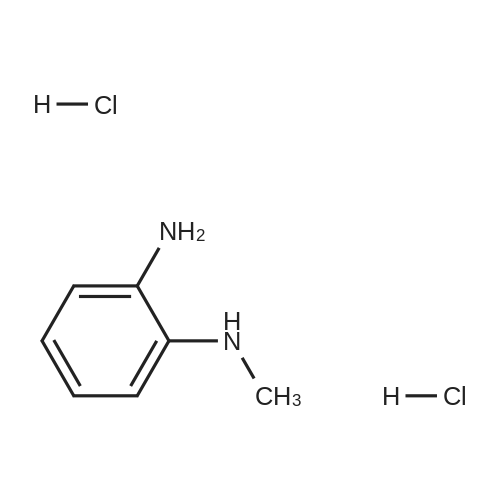
| 60% |
In benzene; at 180℃; for 6h; |
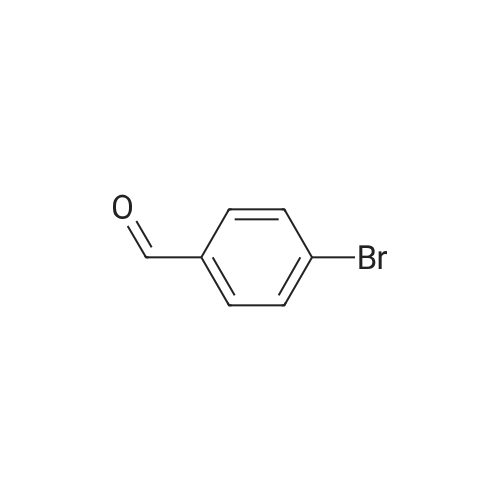
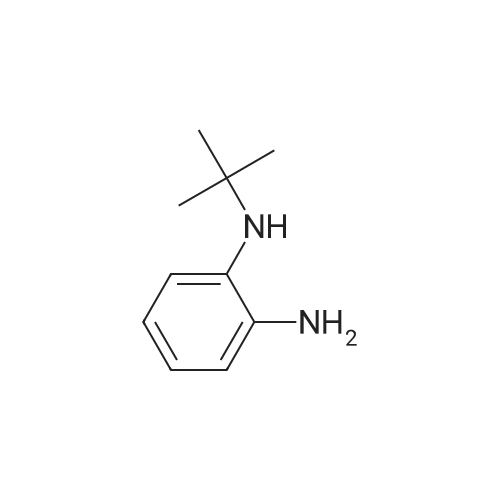
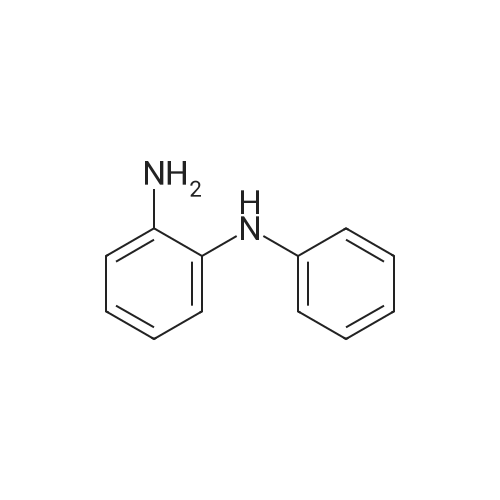
4-bromo-benzaldehyde (8.3g, 45mmol) was dispersed in the benzene in 10ml of N-phenyl-1,2-phenylenediamine (N-phenyl-1,2-phenylenediamine, 8.3g, 45mmol ), which was heated at 180 C for 6 hours. After cooling to room temperature, and then removed by distillation under reduced pressure to nitrobenzene, the resulting solid was filtered and dried in vacuo, washed with ethyl ether to obtain a compound A. |
| 57.71% |
With toluene-4-sulfonic acid; In toluene; for 16h;Heating / reflux; |
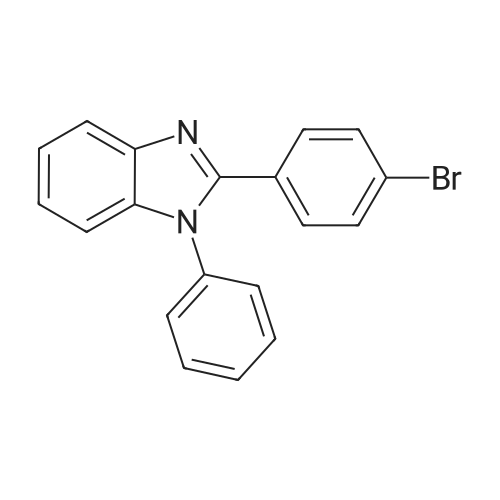
Synthesis of 2-(4-bromophenyl)-1-phenyl-1H-benzo[d]imidazoleN-Phenyl-o-phenylenediamine 13.27 g (72 mmole), 4-bromobenzaldehyde 16 g (87 mmole), and 2.8 g of PTSA (14 mmole) was stirred in 150 ml of Toluene, the reaction mixture was then heated to reflux for 16 hours, after cooling, the reaction mixture was extracted with water, and then the organic layer was evaporated to dry, the residue was then recrystallized with acetone to get 14.51 g of product (yield=57.71%). |
| 49% |
In acetic acid; for 12h;Reflux; |
Acetic acid (20 mL) was added to a flask containing N-phenyl-o-phenylenediamine (1.50 g, 8.14 mmol) and 4-bromobenzaldehyde (1.66 g, 8.96 mmol). After the mixture was refluxed for 12 h, distilled water was added and the organic layer was extracted with dichloromethane. The organic layer was washed with sodium bicarbonate and brine and dried using anhydrous sodium sulfate. The filtrate was concentrated in vacuo to give a crude mixture, which was then subjected to column chromatography on silica gel using ethyl acetate and hexane (v/v 1:20) as the eluent. Analytically pure 2-(4-bromophenyl)-1-phenyl-1H-benz[d]imidazole was isolated as a white solid (1.39 g, 49%). 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 8.4 Hz, 1H), 7.53-7.42 (m, 7 H), 7.35-7.21 (m, 5H). 13C NMR (CDCl3, 100 MHz) δ 151.21, 142.90, 137.25, 136.75, 131.55, 130.84, 130.01, 128.90, 128.77, 127.37, 124.04, 123.58, 123.15, 119.90, 110.48. MALDI-TOF MS: calcd for C19H13BrN2 348.03, found 349.20. |
| 35% |
With Oxone; In N,N-dimethyl-formamide; at 20 - 40℃;Inert atmosphere; |
c) 2-(4-Bromophenyl)-1-phenyl-1H-benzimidazole N-Phenyl-o-phenylenediamine (50 g, 0.27 mol) is dissolved in anhydrous DMF (400 ml) under N2, and 4-bromobenzaldehyde (45.5 g, 0.25 mol) is added dropwise. The reaction mixture is warmed to 40 C., and Oxone (potassium hydrogen monopersulfate, 98.1 g, 0.16 mol) is added in portions. After the mixture has been stirred at room temperature for 120 min., 1 l of water is added. The precipitated product is filtered off, washed with water and dried in vacuo. Recrystallisation from acetonitrile gives a cream-coloured solid (31 g, 35%). |
| 23.6% |
With acetic acid; at 140℃;Inert atmosphere; |
2.1Weigh 7.36 g of 4-bromobenzaldehyde and 7.3 g of o-aminodiphenylamine in 200 mL of acetic acid.After charging and discharging nitrogen for 3 times, the temperature was set to 140 C to start the reaction;2.2After the reaction, the temperature was lowered to room temperature, and a large amount of gray solid was precipitated after pouring into water, and filtered.After the filter cake was added with 5 g of silica gel, the column was passed to obtain 3.2 g of a white solid powder, the yield was 23.6%, HPLC 99.7%; |
|
With Oxone; In N,N-dimethyl-formamide; for 12h;Heating / reflux; |
[Exemplary embodiment 2] Synthesis of compound 3 Compound 3 <n="67"/>[Reaction formula 2]OxoneDMF, reflux 12hr 4-bromobenzaldehyde (5g, 27mmol), Nl-phenylbenzene- 1,2-diamine (5.47g, 29.7mmol) and oxone (18.25g,29.7mmol) were put into a reactor, melted by DMF of 80ml and agitated for 10 hours at 120C to thereby synthesize2- (4-bromophenyl) -1-phenyl-lH-benzo [d] imidazole. The synthesized 2- (4-bromophenyl) -1-phenyl-lH- benzo [d] imidazole (β.98g, 20mmol) was melted by THF of50ml, gradually added with 2Mn-BuLi (11ml, 22mmol) at -78 C and agitated for one hour at low temperatures. The mixture was added with (Z) -3- (5-bromothiophene-2-yl) -2- cyanoacrylic acid (5.1βg, 20mmol) at -78C, agitated for one hour at low temperatures and then additionally agitated for 30 minutes at 0C to complete the reaction. <n="68"/>After extracting an organic layer from the mixture with dichloromethane and distilled water, the solvent was removed through a distiller, the organic layer was recrystallized by n-hexane, and dried and purified after filtering sediment (yield 48%) .1H NMR(CDCl3) : 11.0 (s, IH), 8.16 (s, IH), 7.70 (m, 3H), 7.54 (dd, 3JHH = 8Hz, 4H), 7.3 (s, IH), 7.26 (m, 7H) . |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping