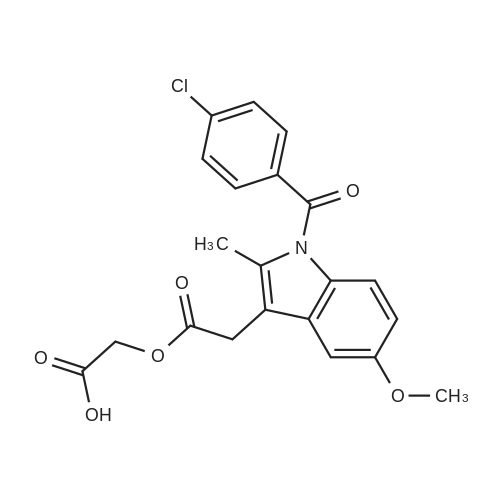
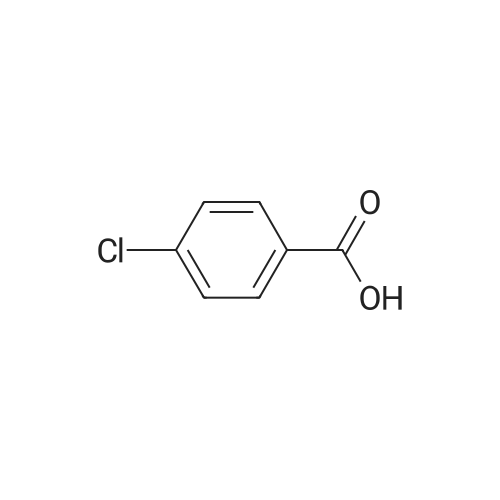
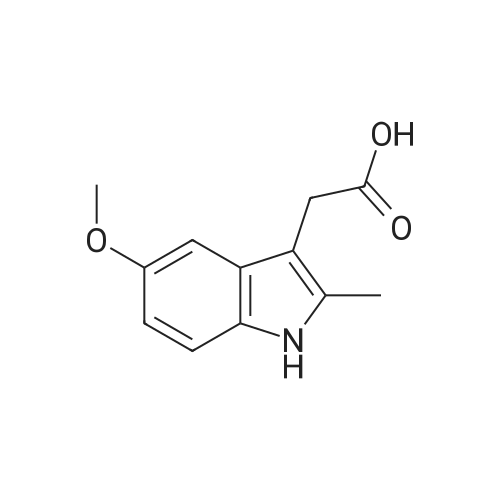
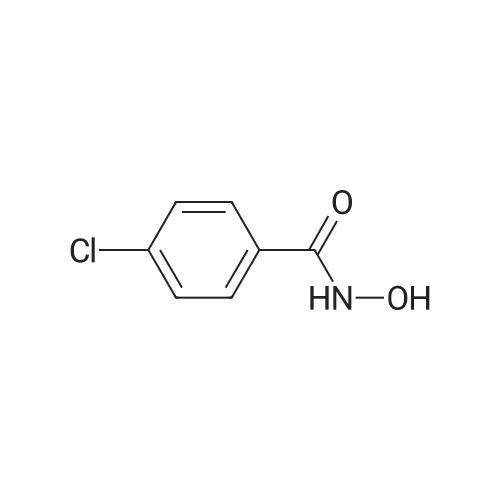
Heterocyclic Building Blocks
Aliphatic Heterocycles(15036)
(1R,5S)-3-azabicyclo[3.1.0]hexane(11)1,2-dithiolane(7)1,4-diazepan-2-one(5)1,4-diazepan-5-one(8)1,4-dioxane(17)1,4-oxazepane(8)10,11-dihydro-5H-dibenzo[b,f]azepine(9)11,12-Didehydro-5,6-dihydrodibenz[b,f]azocine(14)3-((((9H-fluoren-9-yl)methoxy)carbonyl)glycyl)-2,2-dimethyloxazolidine-4-carboxylic acid(8)3H-diazirine(11)4-benzyl-1,4-oxazepane(5)tetrahydro-2H-thiopyran 1,1-dioxide(6)benzazepin(2)benzoxaphosphole(36)
Azetidines(2992)
1-(azetidin-1-yl)-2-bromoethan-1-one(3)1-(azetidin-3-yl)-1H-pyrazole(7)1-(phenylsulfonyl)azetidine(7)1-benzhydrylazetidine(13)1-benzylazetidine(6)1-methylazetidine(6)1-phenylazetidine(8)2,7-diazaspiro[3.5]nonane(7)2-azaspiro[3.3]heptane(11)2-imino-N-((6R,7R)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-7-yl)-2-(thiazol-4-yl)acetamide(3)3-bromoazetidine(3)3-methylazetidin-3-ol(3)3-methyleneazetidine(8)3-phenylazetidine(5)4-(azetidin-1-yl)piperidine(6)azetidin-2-one(10)azetidin-2-ylmethanol(6)azetidin-3-amine(4)azetidin-3-ol(8)azetidin-3-yl acetate(3)azetidin-3-ylmethanamine(3)azetidine-2-carboxylic acid(6)azetidine-3-carboxylic acid(5)benzyl azetidine-1-carboxylate(9)methyl azetidine-2-carboxylate(3)N-((6R,7R)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-7-yl)-2-phenylacetamide(5)tert-butyl azetidine-1-carboxylate(64)
Benzimidazoles(4426)
(1H-benzo[d]imidazol-2-yl)methanamine(6)(1H-benzo[d]imidazol-2-yl)methanol(13)1,2-diphenyl-1H-benzo[d]imidazole(7)1-benzyl-1H-benzo[d]imidazole(6)1-ethyl-1H-benzo[d]imidazole(4)1-methyl-1H-benzo[d]imidazole(37)1-phenyl-1H-benzo[d]imidazole(4)1H-benzo[d]imidazol-2-amine(7)1H-benzo[d]imidazol-5-amine(4)1H-benzo[d]imidazol-6-amine(6)1H-benzo[d]imidazole-2-carbaldehyde(4)1H-benzo[d]imidazole-2-carboxylic acid(7)1H-benzo[d]imidazole-2-thiol(10)1H-benzo[d]imidazole-4-carboxylic acid(6)1H-benzo[d]imidazole-5-carboxylic acid(4)2-((pyridin-2-ylmethyl)thio)-1H-benzo[d]imidazole(7)2-(1H-benzo[d]imidazol-2-yl)ethan-1-amine(5)2-(chloromethyl)-1H-benzo[d]imidazole(9)2-benzyl-1H-benzo[d]imidazole(7)2-bromo-1H-benzo[d]imidazole(3)2-chloro-1H-benzo[d]imidazole(17)2-methyl-1H-benzo[d]imidazole(19)4-bromo-1H-benzo[d]imidazole(10)4-methyl-1H-benzo[d]imidazole(7)5,6-dichloro-1H-benzo[d]imidazole(4)5-bromo-1H-benzo[d]imidazole(9)5-chloro-1H-benzo[d]imidazole(5)5-methoxy-1H-benzo[d]imidazole(2)5-methyl-1H-benzo[d]imidazole(5)6-chloro-1H-benzo[d]imidazole(11)methyl (1H-benzo[d]imidazol-2-yl)carbamate(5)methyl 1H-benzo[d]imidazole-2-carboxylate(3)tert-butyl 1H-benzo[d]imidazole-1-carboxylate(5)benzimidazol-amine(28)benzimidazole-carboxylate(6)benzimidazole-carboxylic acid(43)benzimidazole-hydrochloride(4)benzoimidazol-methanol(4)benzoimidazol-one(50)benzoimidazole-thione(4)bromo-benzimidazole(84)chloro-benzimidazole(5)dimethyl-benzoimidazole(24)dioxaborolane-benzimidazole(3)hydroxy-benzimidazole(23)methylbenzimidazole(2)phenyl-benzoimidazole(75)piperidine-benzimidazole(5)trifluoromethyl-benzimidazole(4)
Benzofurans(3708)
1-(benzofuran-2-yl)ethan-1-one(3)2,2-dimethyl-2,3-dihydrobenzofuran(7)2,3-dihydrobenzofuran-3-amine(11)2,3-dihydrobenzofuran-5-amine(3)2,3-dihydrobenzofuran-7-amine(5)2-(benzofuran-3-yl)acetic acid(3)2-bromodibenzo[b,d]furan(6)2-ethylbenzofuran(3)2-methylbenzofuran(5)3-methylbenzofuran(4)4-bromobenzofuran(3)4-phenyldibenzo[b,d]furan(8)5,7,8-trimethylchromane(3)5-bromo-2,3-dihydrobenzofuran(6)5-chloro-2,3-dihydrobenzofuran(5)5-fluoro-2,3-dihydrobenzofuran(5)5-methoxyisobenzofuran-1(3H)-one(5)6-bromo-2,3-dihydrobenzofuran(7)6-fluoro-2,3-dihydrobenzofuran(6)6-methoxybenzofuran(4)7-bromo-2,3-dihydrobenzofuran(6)7-bromobenzofuran(6)7-fluoro-2,3-dihydrobenzofuran(6)benzofuran-2(3H)-one(7)benzofuran-2-carbaldehyde(5)benzofuran-2-carboxylic acid(21)benzofuran-2-ylboronic acid(8)benzofuran-3(2H)-one(15)benzofuran-3-carboxylic acid(6)benzofuran-3-yl(phenyl)methanone(11)methyl benzofuran-2-carboxylate(6)aminobenzofuran(5)benzofuran-carboxylate(5)benzofuran-carboxylic acid(67)benzofuran-hydrochloride(38)benzofuran-methanol(2)benzofuran-one(89)benzofuranopyrimidine(4)bromobenzofuran(17)bromoisobenzofuran(1)chlorobenzofuran(10)chloroisobenzofuran(10)dihydrobenzofuran(104)dihydroisobenzofuran(11)dimethoxyisobenzofuran(4)epoxyisobenzofuran(4)ethylbenzofuran(3)fluorobenzofuran(19)fluoroisobenzofuran(5)hexahydroisobenzofuran(5)hydroxy-benzofuran(11)hydroxybenzofuran(8)hydroxyisobenzofuran(2)iodo-benzofuran(16)iodoisobenzofuran(2)isobenzofuran(37)methoxybenzofuran(14)methoxyisobenzofuran(4)methylbenzofuran(25)methylbenzofuro(13)methylisobenzofuran(2)nitrobenzofuran(8)nitroisobenzofuran(3)phenyl-benzofuran(4)piperidine-benzofuran(6)tetrahydrobenzofuran(6)
Benzothiazoles(2414)
2-(methylthio)benzo[d]thiazole(6)2-bromobenzo[d]thiazole(28)2-chlorobenzo[d]thiazole(21)2-methylbenzo[d]thiazole(26)4-bromobenzo[d]thiazole(9)4-chlorobenzo[d]thiazole(7)4-fluorobenzo[d]thiazole(6)4-methylbenzo[d]thiazole(10)5-chlorobenzo[d]thiazole(5)5-nitrobenzo[d]thiazole(3)6-bromobenzo[d]thiazole(7)6-chlorobenzo[d]thiazole(6)6-fluorobenzo[d]thiazole(5)6-methoxybenzo[d]thiazole(6)6-methylbenzo[d]thiazole(6)6-nitrobenzo[d]thiazole(3)7-bromobenzo[d]thiazole(8)7-chlorobenzo[d]thiazole(6)7-fluorobenzo[d]thiazole(9)benzo[d]thiazol-2-amine(30)benzo[d]thiazol-5-amine(3)benzo[d]thiazol-6-amine(3)benzo[d]thiazol-6-ol(2)benzo[d]thiazole-2-carbonitrile(11)benzo[d]thiazole-2-thiol(11)benzo[d]thiazole-6-carboxylic acid(5)methyl benzo[d]thiazole-6-carboxylate(5)benzothiazol-amine(6)benzothiazole-carboxylate(18)benzothiazole-carboxylic acid(27)benzothiazole-chloride(41)benzothiazole-nitrile(24)benzothiazole-thione(12)benzothiazolyl-bromide(4)bromobenzothiazole(7)chlorobenzothiazole(4)dichlorobenzothiazole(10)hydroxy-benzothiazole(17)methoxybenzothiazole(4)methylbenzothiazole(7)nitrobenzothiazole(3)trifluoromethyl-benzothiazole(17)
Benzothiophenes(1107)
2-(benzo[b]thiophen-2-yl)-4,5-dihydrooxazole(10)2-methylbenzo[b]thiophene(4)3-bromobenzo[b]thiophene(5)3-chlorobenzo[b]thiophene(6)3-methylbenzo[b]thiophene(4)4,5,6,7-tetrahydrobenzo[b]thiophene(21)5-bromobenzo[b]thiophene(3)5-chlorobenzo[b]thiophene(4)6-bromobenzo[b]thiophene(4)7-bromobenzo[b]thiophene(5)benzo[b]thiophen-2-amine(5)benzo[b]thiophen-2-ylboronic acid(5)benzo[b]thiophene-2-carbaldehyde(6)benzo[b]thiophene-2-carbonitrile(3)benzo[b]thiophene-2-carboxylic acid(32)benzo[b]thiophene-3-carbaldehyde(6)ethyl benzo[b]thiophene-2-carboxylate(9)methyl benzo[b]thiophene-2-carboxylate(27)benzothiophene-amine(4)benzothiophene-bromide(33)benzothiophene-carboxylate(36)benzothiophene-carboxylic acid(58)benzothiophene-dioxaborolane(8)benzothiophene-nitrile(4)benzothiophene-piperazine(5)
Benzoxazoles(2790)
2-chlorobenzo[d]oxazole(11)2-methylbenzo[d]oxazole(26)2-phenylbenzo[d]oxazole(22)3-phenylbenzo[d]oxazol-3-ium(8)5-bromobenzo[d]oxazole(3)5-chlorobenzo[d]oxazole(5)5-methylbenzo[d]oxazole(3)6-chlorobenzo[d]oxazole(4)benzo[d]oxazol-2-amine(16)benzo[d]oxazole-2-thiol(6)methyl benzo[d]oxazole-2-carboxylate(3)amino-benzoxazole(24)benzoxazol-amine(11)benzoxazol-one(3)benzoxazole-carboxylate(3)benzoxazole-carboxylic acid(25)benzoxazole-chloride(4)benzoxazole-methanol(4)dioxaborolane-benzoxazole(3)methyl-benzoxazole(114)phenyl-benzoxazole(4)trifluoromethyl-benzoxazole(1)
Carbazole Series(498)
1-methyl-9H-carbazole(7)2,3,4,9-tetrahydro-1H-carbazol-1-one(7)2-bromo-9H-carbazole(4)3,9-diphenyl-9H-carbazole(7)3-(tert-butyl)-9H-carbazole(3)3-bromo-9-phenyl-9H-carbazole(16)3-bromo-9H-carbazole(4)3-chloro-9H-carbazole(3)3-methyl-9H-carbazole(3)7H-benzo[c]carbazole(5)9-([1,1'-biphenyl]-4-yl)-9H-carbazole(9)9-(p-tolyl)-9H-carbazole(5)9-benzyl-9H-carbazole(7)9-ethyl-9H-carbazole(11)carbazole-carboxylic acid(9)carbazolyl-phenyl-boronic acid(17)carbazolyl-phenyl-boronic acid/ester(6)
Dioxolanes(1577)
(3aS,6aS)-tetrahydrofuro[3,4-d][1,3]dioxole(6)1,3-dioxolan-2-one(11)2,2-difluorobenzo[d][1,3]dioxole(24)2,2-dimethyl-1,3-dioxolane(27)2,2-dimethylbenzo[d][1,3]dioxole(7)2-phenyl-1,3-dioxolane(18)4-bromobenzo[d][1,3]dioxole(6)4-methyl-1,3-dioxolane(5)5,6-dihydro-[1,3]dioxolo[4,5-g]isoquinolino[3,2-a]isoquinolin-7-ium(4)5-bromobenzo[d][1,3]dioxole(12)5-chlorobenzo[d][1,3]dioxole(5)5-nitrobenzo[d][1,3]dioxole(4)9-((3aS,4R,6aR)-tetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purine(7)benzo[d][1,3]dioxol-5-amine(3)benzothiophene-dioxaborolane(8)dioxaborolane-benzyl(3)dioxaborolane-indazole-carboxylate(3)dioxaborolane-indoline(3)dioxaborolane-phenol(44)
Furans(6958)
2,3-dihydrofuran-3-one(9)2-(bromomethyl)furan(3)2-(chloromethyl)furan(3)2-(furan-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(6)2-bromofuran(21)2-chlorofuran(3)2-methylfuran(27)2-nitrofuran(8)3-bromofuran(11)4-methylisobenzofuran-1(3H)-one(3)5-nitro-2,3-dihydrobenzofuran(3)5-phenylfuran-2-carbaldehyde(9)5-phenylfuran-2-carboxylic acid(6)6-chloro-2,3-dihydrobenzofuran(3)6-fluoroisobenzofuran-1(3H)-one(3)7-chloro-2,3-dihydrobenzofuran(3)ethyl furan-2-carboxylate(6)furan-2,5-dione(8)furan-2-carbaldehyde(19)furan-2-carbonitrile(4)furan-2-carboxylic acid(29)furan-2-ylmethanol(7)furan-3-carboxylic acid(9)furo[3,2-b]pyridine(7)furo[3,2-c]pyridine(7)methyl furan-2-carboxylate(14)N-(furan-2-ylmethyl)aniline(5)aminodihydrofuran(7)aminofuran(6)aminofuro(3)Bromo-naphtho-benzofuran(2)bromofuran(32)chlorofuran(2)chlorofuro(11)cyanofuran(54)dibromofuran(5)dichlorofuro(3)dihydrofuran(31)dihydrofuro(15)dihydroxydihydrofuran(8)dimethylfuran(10)formylfuran(10)furan-one(251)furanopyrimidine(12)furochromen(7)furopyridine(31)furopyrrole(6)hexahydrofuro(6)hydroxydihydrofuran(34)methyldihydrofuran(11)methylfuran(58)nitrofuran(10)trifluoromethyl-furan(23)
Imidazoles(4790)
1,1'-diphenyl-4,4',5,5'-tetrahydro-1H,1'H-2,2'-biimidazole(11)1,2-dimethyl-1H-imidazole(6)1,3-diphenyl-1H-imidazol-3-ium(14)1,3-diphenyl-4,5-dihydro-1H-imidazol-3-ium(5)1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one(3)1-(phenylsulfonyl)-1H-imidazole(5)1-benzyl-1H-imidazole(20)1-decyl-3-methyl-1H-imidazol-3-ium(14)1-ethyl-1H-imidazole(3)1-isopropyl-1H-imidazole(5)1-phenyl-1H-imidazole-4-carboxylic acid(4)1-trityl-1H-imidazole(15)1H-imidazo[4,5-b]pyridine(14)1H-imidazo[4,5-c]pyridine(12)1H-imidazol-2-amine(6)1H-imidazole-2-carboxylic acid(7)1H-imidazole-4-carbonitrile(2)1H-imidazole-4-carboxylic acid(4)2-(pyridin-2-yl)-1H-benzo[d]imidazole(5)2-bromo-1H-imidazole(13)2-bromoimidazo[1,2-a]pyridine(5)2-ethyl-1H-imidazole(3)2-methyl-1-phenyl-1H-imidazole(10)2-methylimidazo[1,2-a]pyridine(8)2-phenyl-1H-benzo[d]imidazole(26)2-phenyl-1H-imidazole(24)2-phenylimidazo[1,2-a]pyridine(12)2-propyl-1H-imidazole(5)3-bromoimidazo[1,2-a]pyrazine(6)3-bromoimidazo[1,2-a]pyridine(26)3-bromoimidazo[1,2-a]pyrimidine(6)3-iodoimidazo[1,2-a]pyridine(10)3-methylimidazo[1,2-a]pyridine(3)4,5,6,7-tetrahydro-3H-imidazo[4,5-c]pyridine(5)4-(1H-imidazol-1-yl)benzaldehyde(6)4-bromo-1-phenyl-1H-imidazole(9)4-methyl-1-phenyl-1H-imidazole(6)4-methyl-1H-imidazole(6)4-phenyl-1H-imidazole(8)5,6,7,8-tetrahydroimidazo[1,2-a]pyrazine(15)5,6,7,8-tetrahydroimidazo[1,2-a]pyridine(12)5,6,7,8-tetrahydroimidazo[1,5-a]pyrazine(11)5-benzyl-1H-imidazole(1)5-bromoimidazo[1,2-a]pyridine(6)5-chloro-1H-imidazole(2)5-fluoro-1H-benzo[d]imidazole(7)5-iodo-1H-imidazole(4)5-methylimidazo[1,2-a]pyridine(4)5-phenyl-1H-imidazole(6)6,7-dihydro-5H-pyrrolo[1,2-a]imidazole(9)6-bromoimidazo[1,2-a]pyrazine(11)6-bromoimidazo[1,2-a]pyridine(14)6-bromoimidazo[1,2-a]pyrimidine(5)6-chloroimidazo[1,2-a]pyridine(4)6-chloroimidazo[1,2-b]pyridazine(30)7-methylimidazo[1,2-a]pyridine(3)8-bromoimidazo[1,2-a]pyrazine(7)8-bromoimidazo[1,2-a]pyridine(15)8-chloroimidazo[1,2-a]pyrazine(8)8-chloroimidazo[1,2-a]pyridine(7)8-fluoroimidazo[1,2-a]pyridine(3)8-methylimidazo[1,2-a]pyridine(9)ethyl 1H-imidazole-2-carboxylate(6)ethyl 2-(imidazo[1,2-a]pyridin-3-yl)acetate(3)ethyl imidazo[1,2-a]pyridine-2-carboxylate(25)ethyl imidazo[1,2-a]pyridine-3-carboxylate(13)imidazo[1,2-a]pyridin-3-amine(7)imidazo[1,2-a]pyridin-8-amine(7)imidazo[1,2-a]pyridin-8-ol(3)imidazo[1,2-a]pyridine-2-carbaldehyde(8)imidazo[1,2-a]pyridine-2-carboxylic acid(21)imidazo[1,2-a]pyridine-3-carbaldehyde(8)imidazo[1,2-a]pyridine-3-carboxylic acid(14)imidazo[1,2-a]pyridine-7-carboxylic acid(3)imidazo[1,2-a]pyridine-8-carboxylic acid(6)imidazo[1,2-a]pyrimidine-3-carboxylic acid(5)imidazo[1,2-c]pyrimidine(6)imidazo[1,5-a]pyrazine(9)imidazo[2,1-b]thiazole(14)methyl 1H-imidazole-2-carboxylate(4)methyl 1H-imidazole-5-carboxylate(5)methyl imidazo[1,2-a]pyridine-8-carboxylate(3)tert-butyl 1H-imidazole-1-carboxylate(6)amino-imidazole(82)aminoimidazole-carboxylate(23)benzyl-methyl-imidazole(2)biimidazole(5)bromo-imidazole(91)bromo-imidazopyridine(85)bromo-methyl-imidazole(88)bromoimidazo(102)bromoimidazole carboxylate(23)chloro-imidazopyridine(78)chloroimidazo(62)dibromoimidazo(8)dichloroimidazo(9)dihydroimidazo(25)dimethyl-imidazol(78)dimethylimidazo(9)dimethylimidazolium(6)fluoroimidazo(24)hydroxy-imidazopyridine(3)imidazol-amine(95)imidazol-ethanol(13)imidazol-hydrochloride(63)imidazol-methanol(22)imidazol-pyridine(98)imidazole-carboxylate(69)imidazole-carboxylic acid(53)imidazolyl-ethanol(3)imidazolyl-nitrile(37)imidazolyl-pyrimidine(2)imidazopyrazine(99)imidazopyridazine(62)imidazopyridin-amine(40)imidazopyridin-one(25)imidazopyridine(627)imidazopyridine-carbonitrile(7)imidazopyridine-carboxylate(77)imidazopyridine-carboxylic acid(66)imidazopyrimidine(84)imidazoquinoline(2)imidazothiazole(42)iodoimidazo(19)iodomethyl-imidazole(9)methoxyimidazo(13)methyl-imidazopyridine(84)methyl-nitro-imidazole(44)methylimidazo(121)methylimidazole(5)methylimidazolium(30)nitro-imidazole-carboxylate(6)nitroimidazole(19)phenylimidazo(7)phenylimidazole(8)piperidin-imidazolyl(39)pyrroloimidazole(13)tetrahydroimidazo(40)trifluoromethyl-imidazole(35)trifluoromethyl-imidazopyridine(30)
Imidazolidines(1145)
(3aR,6aS)-tetrahydro-1H-thieno[3,4-d]imidazol-2(3H)-one(18)1-phenylimidazolidin-2-one(8)3-phenyl-2-thioxoimidazolidin-4-one(6)3-phenylimidazolidine-2,4-dione(4)imidazolidin-2-one(14)imidazolidine-2,4-dione(17)benzylimidazolidine(8)dioxoimidazolidin(3)imidazolidin-one(34)methylimidazolidine(3)oxoimidazolidine(8)thioxoimidazolidin(9)
Indazoles(3077)
(1H-indazol-4-yl)boronic acid(3)(1H-indazol-6-yl)boronic acid(4)1-(1H-indazol-1-yl)ethan-1-one(7)1-(tetrahydro-2H-pyran-2-yl)-1H-indazole(17)1-benzyl-1H-indazole(7)1-ethyl-1H-indazole(7)1H-indazol-3-amine(23)1H-indazol-3-ol(10)1H-indazol-4-amine(12)1H-indazol-4-ol(4)1H-indazol-5-amine(3)1H-indazol-6-amine(5)1H-indazol-6-ol(6)1H-indazol-7-amine(6)1H-indazole-3-carbaldehyde(19)1H-indazole-3-carbonitrile(9)1H-indazole-3-carboxylic acid(24)1H-indazole-4-carboxylic acid(8)1H-indazole-5-carboxylic acid(4)1H-indazole-7-carboxylic acid(4)2,3-dimethyl-2H-indazole(4)2-methyl-2H-indazole(34)3-(piperidin-4-yl)-1H-indazole(5)3-chloro-1H-indazole(13)4,5,6,7-tetrahydro-1H-indazole(12)4-chloro-1H-indazole(18)4-fluoro-1H-indazole(22)4-methoxy-1H-indazole(7)4-methyl-1H-indazole(12)4-nitro-1H-indazole(10)5-bromo-1H-indazole(14)5-chloro-1H-indazole(9)5-methoxy-1H-indazole(5)5-nitro-1H-indazole(5)6-(trifluoromethyl)-1H-indazole(6)6-bromo-1H-indazole(23)6-chloro-1H-indazole(14)6-fluoro-1H-indazole(15)6-methoxy-1H-indazole(14)6-methyl-1H-indazole(9)6-nitro-1H-indazole(9)7-bromo-1H-indazole(18)7-chloro-1H-indazole(7)7-fluoro-1H-indazole(12)7-methoxy-1H-indazole(5)7-nitro-1H-indazole(11)methyl 1H-indazole-3-carboxylate(23)methyl 1H-indazole-4-carboxylate(10)methyl 1H-indazole-5-carboxylate(3)methyl 1H-indazole-6-carboxylate(9)methyl 1H-indazole-7-carboxylate(8)methyl 2-methyl-2H-indazole-3-carboxylate(3)tert-butyl 1H-indazole-1-carboxylate(29)amino-indazole(21)amino-indazole-carboxylate(12)bromo-fluoro-indazole(43)bromo-indazol-amine(4)bromo-indazole(173)bromo-indazole-carboxylate(26)bromo-methoxy-indazole(13)bromo-nitro-indazole(12)bromomethyl-indazole(23)chlorindazole-carboxylic-acid(6)chloro-indazole(44)chloro-indazole-amine(1)chloro-indazole-carboxylate(3)chloro-methyl-indazole(27)dimethyl-indazole(13)dioxaborolan-indazole(27)dioxaborolane-indazole-carboxylate(3)fluoro-indazol-amine(24)fluoro-indazole-carboxylate(20)fluoro-indazole-carboxylic-acid(3)fluoro-methyl-indazole(74)indazol-amide(3)indazol-amine(89)indazol-boronic acid(36)indazol-one(8)indazole-benzene(30)indazole-carbonitrile(25)indazole-carboxylate(36)indazole-carboxylic acid(66)indazole-hydrochloride(2)indazole-hydroxyl(10)indazole-methanol(6)iodo-indazol-amine(3)iodo-indazole(65)iodo-indazole-carboxylate(4)iodomethyl-indazole(24)methoxy-indazole-carboxylate(1)methoxyindazole-carboxylic-acid(2)methyl-indazolamide(13)methyl-nitro-indazole(25)methylindazole(6)nitro-indazol-amine(3)tetrahydropyran-indazole(4)trifluoromethyl-indazole(11)
Indoles(7651)
(1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid(17)(1H-indol-2-yl)boronic acid(3)(1H-indol-2-yl)methanol(7)(3aS,7aS)-octahydro-1H-indole(5)(9H-fluoren-9-yl)methyl (2-(1H-indol-3-yl)ethyl)carbamate(12)1,2,3,4-tetrahydrocyclopenta[b]indole(5)1,2-dimethyl-1H-indole(4)1-(1H-indol-1-yl)ethan-1-one(9)1-(1H-indol-3-yl)ethan-1-one(6)1-(phenylsulfonyl)-1H-indole(26)1-(triisopropylsilyl)-1H-pyrrolo[2,3-b]pyridine(8)1-benzyl-1H-indole(10)1-ethyl-1H-indole(7)1-methyl-1H-indole(44)1-methyl-1H-indole-2-carboxylic acid(8)1-methyl-1H-indole-3-carbaldehyde(9)1-methyl-1H-pyrrolo[2,3-b]pyridine(14)1-phenyl-1H-indole(5)1H-indol-3-ol(5)1H-indol-3-yl acetate(5)1H-indol-4-amine(9)1H-indol-4-ol(7)1H-indol-5-ol(4)1H-indol-6-amine(14)1H-indol-6-ol(6)1H-indol-7-amine(8)1H-indole-2-carbaldehyde(18)1H-indole-2-carbonitrile(9)1H-indole-3-carbonitrile(17)1H-indole-3-carboxylic acid(26)1H-indole-4-carboxylic acid(9)1H-indole-5-carbonitrile(5)1H-indole-5-carboxylic acid(4)1H-indole-6-carbonitrile(4)1H-indole-6-carboxylic acid(5)1H-indole-7-carbonitrile(6)1H-indole-7-carboxylic acid(7)1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde(7)1H-pyrrolo[2,3-b]pyridine-5-carbaldehyde(5)2,3,4,5-tetrahydro-1H-pyrido[4,3-b]indole(12)2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole(13)2,3-dimethyl-1H-indole(9)2-(1,3,2-dioxaborolan-2-yl)-1-methyl-1H-indole(6)2-(1H-indol-1-yl)acetic acid(5)2-(1H-indol-3-yl)acetic acid(23)2-(1H-indol-3-yl)ethan-1-amine(4)2-(1H-indol-3-yl)ethan-1-ol(10)2-(2-methyl-1H-indol-3-yl)acetic acid(5)2-(4-fluorophenyl)-1H-indole(4)2-methyl-1H-indole-3-carbaldehyde(3)2-methyl-1H-pyrrolo[2,3-b]pyridine(12)3-(1-oxoisoindolin-2-yl)piperidine-2,6-dione(21)3-(1H-indol-3-yl)propanoic acid(4)3-(piperidin-4-yl)-1H-indole(8)3-iodo-1H-pyrrolo[2,3-b]pyridine(11)3-methyl-1H-indole(25)3H-indole(8)4-bromo-1H-indole(25)4-chloro-1H-indole(22)4-fluoro-1H-indole(24)4-fluoro-1H-pyrrolo[2,3-b]pyridine(6)4-fluoroindoline-2,3-dione(3)4-methoxy-1H-indole(11)4-methyl-1H-indole(13)4-nitro-1H-indole(6)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole(5)5-(benzyloxy)-1H-indole(8)5-bromo-1-methyl-1H-indole(4)5-chloro-1H-indole(29)5-chloro-1H-indole-2-carboxylic acid(6)5-chloro-1H-pyrrolo[2,3-b]pyridine(13)5-fluoro-1H-indole(21)5-fluoro-1H-pyrrolo[2,3-b]pyridine(9)5-fluoroindolin-2-one(4)5-methoxy-1H-indole(11)5-methoxy-2-methyl-1H-indole(5)5-methyl-1H-indole(17)5-nitro-1H-indole(6)6-(trifluoromethyl)-1H-indole(5)6-bromo-1-methyl-1H-indole(3)6-bromo-1H-indole(15)6-bromo-1H-pyrrolo[2,3-b]pyridine(6)6-chloro-1H-indole(19)6-chloro-1H-pyrrolo[2,3-b]pyridine(13)6-fluoro-1H-indole(17)6-methoxy-1H-indole(12)6-methyl-1H-indole(17)6-methyl-1H-pyrrolo[2,3-b]pyridine(8)6-nitro-1H-indole(8)7-bromo-1H-indole(24)7-bromoindoline-2,3-dione(3)7-chloro-1H-indole(21)7-fluoro-1H-indole(18)7-fluoro-1H-indole-2-carboxylic acid(5)7-methoxy-1H-indole(8)7-methyl-1H-indole(30)7-nitro-1H-indole(11)9H-pyrido[3,4-b]indole(5)benzo[cd]indol-2(1H)-one(5)ethyl 1H-indole-3-carboxylate(6)ethyl 5-chloro-1H-indole-2-carboxylate(5)ethyl 5-fluoro-1H-indole-2-carboxylate(5)ethyl 6-chloro-1H-indole-2-carboxylate(5)ethyl 7-chloro-1H-indole-2-carboxylate(5)methyl 1H-indole-3-carboxylate(21)methyl 1H-indole-4-carboxylate(9)methyl 1H-indole-5-carboxylate(7)methyl 1H-indole-6-carboxylate(9)methyl 1H-indole-7-carboxylate(7)methyl 2-(1H-indol-3-yl)acetate(3)N-(2-(1H-indol-3-yl)ethyl)acetamide(5)spiro[indoline-3,4'-piperidin]-2-one(18)tert-butyl 1H-indole-1-carboxylate(18)tert-butyl 1H-pyrrolo[2,3-b]pyridine-1-carboxylate(9)tert-butyl 2-(1,3,2-dioxaborolan-2-yl)-1H-indole-1-carboxylate(6)tert-butyl 3-(hydroxymethyl)-1H-indole-1-carboxylate(10)tert-butyl 3-bromo-1H-indole-1-carboxylate(9)tert-butyl 3-iodo-1H-indole-1-carboxylate(7)tert-butyl 5-methoxy-1H-indole-1-carboxylate(6)tryptophan(23)aminoindole(6)azaindole(44)benzoindole(11)benzyloxyindole(1)biindolinylidene(1)bromo-indole-carboxylate(56)bromo-indole-carboxylic acid(28)bromo-methyl-indole(3)bromoindole(8)chlorindole-carboxylic-acid(2)chloro-indole-carboxylate(41)chloro-methyl indole(111)chloroindole(6)cyanoindole(5)dihydroindolo(14)dimethyl-indole(105)dioxaborolan-indole(90)fluorindole-carboxylic acid(26)fluorindole-carboxylic-acid(4)fluoro-indole-carboxylate(54)fluoroindole(5)fluoromethyl-indole(102)formyl-indole-carboxylate(15)formylindole(44)hydroxyindole(7)hydroxyisoindoline(2)hydroxymethy-indole-carboxylate(2)indol-amine(85)indol-methanamine(17)indol-methanol(8)indol-one(249)indole-boronic acid(82)indole-carbonitrile(33)indole-ethanol(6)indole-ethylamine(3)indole-hydrochloride(94)indole-hydroxyl(138)indolecarboxylate(2)indolecarboxylic(206)iodo-indole(46)isoindole(26)methoxy-indole-carboxylate(27)methoxy-methyl-indole(87)methoxyindole(9)methyl-nitro-indole(5)methylindole(16)nitro-indole-carboxylate(18)nitroindole(6)phenyl-indole(163)piperidine-indole(105)pyridinoindole(25)pyrimido-indole(5)trifluoromethyl-indole(47)
Indolines(4462)
(E)-[3,3'-biindolinylidene]-2,2'-dione(5)(Z)-3-((1H-pyrrol-2-yl)methylene)indolin-2-one(8)(Z)-3-(phenyl(phenylamino)methylene)indolin-2-one(5)(Z)-3-benzylideneindolin-2-one(7)1-(indolin-1-yl)ethan-1-one(9)1-benzylindoline-2,3-dione(5)1-methylindolin-2-one(14)1-methylindoline(3)1-methylindoline-2,3-dione(8)2-(1,3-dioxoisoindolin-2-yl)propanoic acid(3)2-methylindoline(7)2-methylisoindolin-1-one(7)3,3-dimethylindolin-2-one(5)3,3-dimethylindoline(7)4-bromoindoline(4)4-chloroindoline-2,3-dione(6)4-methylindoline(3)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indoline(5)5-bromoindolin-2-one(5)5-bromoindoline(8)5-bromoisoindoline-1,3-dione(3)5-chloroindolin-2-one(3)5-fluoroindoline(3)5-methoxyindoline(4)5-methylindolin-2-one(3)5-methylindoline(3)6-bromoindolin-2-one(8)6-bromoindoline-2,3-dione(4)6-chloroindolin-2-one(5)6-chloroindoline(3)6-chloroindoline-2,3-dione(5)6-fluoroindolin-2-one(5)6-fluoroindoline-2,3-dione(3)7-bromoindoline(3)7-fluoroindolin-2-one(3)7-fluoroindoline-2,3-dione(6)7-methylindoline-2,3-dione(7)indoline-2-carboxylic acid(3)methyl indoline-6-carboxylate(3)spiro[cyclopropane-1,3'-indolin]-2'-one(6)tert-butyl indoline-1-carboxylate(24)aminoindoline(10)aminoisoindoline(6)bromoindoline(17)bromoisoindolin(5)bromoisoindoline(6)chloroindolin(4)chloroisoindoline(3)dibromoindoline(12)dichloroindoline(16)difluoroindoline(1)dimethylindoline(21)dioxaborolane-indoline(3)dioxoindoline(8)dioxoisoindolin(72)fluoroindolin(9)fluoroindoline(9)fluoroisoindoline(10)hydroxy-indoline(3)hydroxyindolin(5)hydroxyisoindolin(40)indolin-one(189)indoline-carboxylate(59)indoline-carboxylic acid(19)indoline-nitrile(4)indoline-oxazine(4)isoindolin(14)isoindoline(20)methoxyindoline(12)methoxyisoindoline(9)methylindoline(46)methylisoindoline(18)nitroindolin(6)nitroindoline(11)nitroisoindoline(8)oxoindoline(32)oxoisoindoline(16)spiro-indoline-piperidine(34)trifluoromethyl-indoline(18)
Isoquinolines(2690)
1,3-dichloroisoquinoline(14)1-bromoisoquinoline(20)1-chloroisoquinoline(20)1-methylisoquinoline(9)1H-benzo[de]isoquinoline-1,3(2H)-dione(14)2-methyl-3,4-dihydroisoquinolin-1(2H)-one(6)3-bromoisoquinoline(10)3-chloroisoquinoline(20)3-methylisoquinoline(8)4-bromoisoquinolin-1(2H)-one(3)4-bromoisoquinoline(33)4-chloroisoquinoline(11)4-fluoroisoquinoline(6)5,6-dihydro-[1,3]dioxolo[4,5-g]isoquinolino[3,2-a]isoquinolin-7-ium(4)5-bromoisoquinoline(17)5-chloroisoquinoline(6)5-methoxyisoquinoline(6)5-nitroisoquinoline(7)6-bromo-3,4-dihydroisoquinolin-1(2H)-one(4)6-bromoisoquinoline(9)6-methoxy-3,4-dihydroisoquinolin-1(2H)-one(3)7-bromo-3,4-dihydroisoquinolin-1(2H)-one(5)7-bromoisoquinoline(6)7-chloroisoquinoline(6)7-fluoroisoquinoline(3)7-methoxyisoquinoline(5)8-bromoisoquinoline(17)8-chloroisoquinoline(7)8-fluoroisoquinolin-1(2H)-one(3)8-fluoroisoquinoline(10)8-methoxyisoquinoline(7)isoquinolin-1-amine(18)isoquinolin-1-ol(8)isoquinolin-3-amine(13)isoquinolin-3-ol(6)isoquinolin-4-amine(16)isoquinolin-4-ol(9)isoquinolin-4-ylboronic acid(3)isoquinolin-5-amine(9)isoquinolin-5-ol(7)isoquinolin-8-amine(6)isoquinoline-1-carbonitrile(7)isoquinoline-1-carboxylic acid(10)isoquinoline-3-carboxylic acid(7)isoquinoline-4-carboxylic acid(12)isoquinoline-5-carboxylic acid(5)isoquinoline-5-sulfonyl chloride(3)isoquinoline-6-carboxylic acid(3)methyl isoquinoline-3-carboxylate(3)methyl isoquinoline-4-carboxylate(6)tert-butyl 3,4-dihydroisoquinoline-2(1H)-carboxylate(19)aminoisoquinoline(13)aminoisoquinoline-carboxylic-acid(4)bromoisoquinoline(69)chloroisoquinoline(101)dibromoisoquinoline(9)dichloroisoquinoline(14)dihydroisoquinoline(107)ethynylisoquinoline(2)fluoroisoquinoline(78)hydroxyisoquinoline(12)iodo-isoquinoline(12)Isoquinolin-boronic-acid(5)isoquinolin-one(105)isoquinoline-benzene(27)isoquinoline-hydrochloride(7)isoquinoline-methanol(7)isoquinolinecarboxylate(4)isoquinolinecarboxylic(2)methoxyisoquinoline(59)methylisoquinoline(59)nitroisoquinoline(23)phenoxyisoquinoline(3)trifluoromethyl-isoquinoline(7)
Isoxazoles(3402)
(3-phenylisoxazol-5-yl)methanol(7)3-bromoisoxazole(7)3-methyl-5-phenylisoxazole(6)3-methylisoxazole(30)3-phenylisoxazol-5-amine(8)5-(thiophen-2-yl)isoxazole(5)5-cyclopropylisoxazole(5)5-methyl-3-phenylisoxazole(4)5-methylisoxazole(16)5-phenylisoxazole-3-carboxylic acid(15)ethyl isoxazole-3-carboxylate(8)ethyl isoxazole-4-carboxylate(5)isoxazol-3-amine(7)isoxazol-3-ylmethanol(3)isoxazol-5-amine(15)isoxazol-5-ylmethanol(5)isoxazole-3-carboxylic acid(8)isoxazole-5-carboxylic acid(7)methyl 5-phenylisoxazole-3-carboxylate(6)N-((5R,6R)-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-6-yl)-3-phenylisoxazole-4-carboxamide(1)aminoisoxazole(7)bromoisoxazole(5)dihydroisoxazol(2)dimethylisoxazole(17)ethylisoxazol(2)isoxazole(2)isoxazole-carboxylate(45)isoxazolecarboxylic(48)methylisoxazole(52)phenylisoxazole(28)
Morpholines(4974)
(R)-3-phenylmorpholine(7)(S)-3-phenylmorpholine(10)2,2-dimethylmorpholine(5)2,6-dimethylmorpholine(5)2-(methoxymethyl)morpholine(4)2-(morpholin-2-yl)acetic acid(5)2-ethylmorpholine(3)2-isopropylmorpholine(4)2-methylmorpholine(7)2-phenylmorpholine(8)3-(4-morpholinophenyl)oxazolidin-2-one(5)3-(methoxymethyl)morpholine(4)3-ethylmorpholine(4)3-isopropylmorpholine(5)3-methylmorpholine(13)3-phenylmorpholine(8)4-(2-fluorophenyl)morpholine(8)4-(3-phenoxypropyl)morpholine(7)4-(o-tolyl)morpholine(6)4-(pyrazin-2-yl)morpholine(5)4-(pyridin-4-yl)morpholine(5)4-(pyrimidin-2-yl)morpholine(9)4-benzylmorpholin-3-one(6)4-morpholinoaniline(4)4-morpholinobenzaldehyde(8)4-morpholinobenzoic acid(5)4-phenylmorpholin-3-one(6)6,6-dimethylmorpholine-3-carboxylic acid(5)benzyl morpholine-4-carboxylate(5)ethyl morpholine-2-carboxylate(4)methyl 2-(morpholin-3-yl)acetate(3)methyl morpholine-2-carboxylate(4)methyl morpholine-3-carboxylate(10)morpholin-2-ylmethanamine(8)morpholin-2-ylmethanol(12)morpholin-3-ylmethanol(14)morpholine-2-carbonitrile(4)morpholine-2-carboxylic acid(12)morpholine-3-carboxylic acid(10)morpholino(phenyl)methanone(11)tert-butyl (morpholin-2-ylmethyl)carbamate(3)tert-butyl 2-(bromomethyl)morpholine-4-carboxylate(3)tert-butyl morpholine-4-carboxylate(23)benzylmorpholine(25)cyanomorpholine(6)dimorpholine(3)dioxaborolane-morpholine(33)diphenylmorpholine(3)ethylmorpholine(9)hydroxyethyl-morpholine(3)hydroxymethylmorpholine(12)isopropylmorpholine(9)methylmorpholine(68)morpholin-hydrochloride(125)morpholin-methanol(21)morpholine-carboxylic acid(43)morpholine-nitrile(26)morpholinecarboxylate(10)morpholinobenzaldehyde(11)morpholinobenzo(13)morpholinobenzoic(8)morpholinobenzonitrile(10)morpholinoethanamine(3)morpholinoethanone(5)morpholinoethoxy(1)morpholinoethyl(5)morpholinomethyl(16)morpholinone(4)morpholinophenyl(14)morpholinopropoxy(6)morpholinopyridine(11)morpholinopyrimidin(6)morpholinosulfonyl(7)oxomorpholine(6)phenylmorpholine(15)pyrazo-morpholine(16)pyrimidine-morpholine(65)trifluoromethyl-phenyl-morpholine(13)
Naphthyridines(946)
1,5-naphthyridine-3-carboxylic acid(6)1,6-naphthyridine(22)1,7-naphthyridine(23)2,7-naphthyridine(5)3-bromo-1,5-naphthyridine(8)4-bromo-1,5-naphthyridine(5)4-chloro-1,5-naphthyridine(6)5,6,7,8-tetrahydro-1,6-naphthyridine(22)5,6,7,8-tetrahydro-1,7-naphthyridine(6)bromo-naphthyridin-one(5)bromo-naphthyridine(61)chloro-naphthyridin-one(5)chloro-naphthyridine(76)fluoro-naphthyridine(2)hydroxy-naphthyridine(16)methoxy-naphthyridine(6)methyl-naphthyridine(55)naphthyridin-amine(19)naphthyridin-one(56)naphthyridine hydrochloride(19)naphthyridine-carboxylate(34)naphthyridine-carboxylic acid(33)tetrahydronaphthyridin(20)
Oxazines(239)
2,2-dimethyl-2H-benzo[b][1,4]oxazin-3(4H)-one(3)2-methyl-2H-benzo[b][1,4]oxazin-3(4H)-one(3)2H-pyrido[3,2-b][1,4]oxazin-3(4H)-one(8)3,4-dihydro-2H-pyrido[3,2-b][1,4]oxazine(5)6-amino-2H-benzo[b][1,4]oxazin-3(4H)-one(3)6-bromo-2H-benzo[b][1,4]oxazin-3(4H)-one(6)6-bromo-3,4-dihydro-2H-benzo[b][1,4]oxazine(5)6-chloro-2H-benzo[b][1,4]oxazin-3(4H)-one(8)6-chloro-3,4-dihydro-2H-benzo[b][1,4]oxazine(5)7-bromo-3,4-dihydro-2H-benzo[b][1,4]oxazine(7)spiro[benzo[d][1,3]oxazine-4,4'-piperidin]-2(1H)-one(5)amino-benzooxazine(4)benzoxazin-one(50)benzoxazine(11)benzoxazine-carboxylate(9)bromo-benzooxazin-one(11)dimethyl-pyridooxazin(24)hydroxy-benzooxazine(2)indenotriazolooxazine(5)indoline-oxazine(4)methy-pyrazolooxazine(4)methyl-benzooxazine(4)nitro-benzooxazin(9)phenoxazine(7)pyrazolooxazine(17)pyridooxazin-one(1)pyridoxazine(37)triazol-oxazine(5)
Oxazoles(2023)
2-bromooxazole(7)2-chlorooxazole(7)2-methyloxazole(19)2-phenyloxazole(16)3,4-diphenylisoxazole(8)4,5-diphenyloxazole(7)4-(oxazol-5-yl)aniline(3)4-methyloxazole(9)5-(4-nitrophenyl)oxazole(3)5-methyloxazole(5)6-bromobenzo[d]oxazole(3)ethyl oxazole-4-carboxylate(8)ethyl oxazole-5-carboxylate(3)methyl oxazole-4-carboxylate(9)oxazol-2-amine(8)oxazole-4-carboxylic acid(4)oxazolo[4,5-b]pyridine(7)oxazolo[5,4-b]pyridine(5)aminooxazole(3)bioxazole(12)bromooxazole(6)bromooxazolo(3)chlorooxazole(5)chlorooxazolo(8)dimethyloxazole(6)diphenyloxazole(7)indenoxazole(58)methyloxazole(31)methyloxazolo(4)oxazole-carboxylate(105)oxazole-carboxylic acid(110)oxazolooxazole(2)phenyloxazole(16)trifluoromethyl-oxazole(61)
Oxazolidines(1300)
(R)-4-benzyloxazolidin-2-one(5)(S)-4-benzyloxazolidin-2-one(11)3-(4-morpholinophenyl)oxazolidin-2-one(5)3-phenyloxazolidin-2-one(4)4-isopropyloxazolidine(6)oxazolidine(21)oxazolidine-2,5-dione(9)benzyloxazolidine(1)dimethyloxazolidin(6)dimethyloxazolidine(8)dioxooxazolidin(2)isopropyloxazolidine(1)methyloxazolidin(7)oxazolidinone(5)oxazolopyrazin-one-hydrochloride(3)phenyloxazolidin(9)phenyloxazolidine(2)propionyloxazolidin(5)
Oxazolines(1579)
(3aR,8aS)-2-(pyridin-2-yl)-3a,8a-dihydro-8H-indeno[1,2-d]oxazole(6)(3aS,8aR)-2-(pyridin-2-yl)-3a,8a-dihydro-8H-indeno[1,2-d]oxazole(7)(4R,5S)-4,5-diphenyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(5)(R)-4-benzyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(7)(R)-4-phenyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(7)(S)-4-benzyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(8)(S)-4-phenyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(6)1,3-bis(4,5-dihydrooxazol-2-yl)benzene(8)2,2'-(1,3-diphenylpropane-2,2-diyl)bis(4,5-dihydrooxazole)(5)2,2'-(2-phenylethane-1,1-diyl)bis(4,5-dihydrooxazole)(5)2,3-dihydropyrazolo[5,1-b]oxazole(7)2,6-bis(4,5-dihydrooxazol-2-yl)pyridine(12)2-(6-benzhydrylpyridin-2-yl)-4,5-dihydrooxazole(10)2-(6-cyclopropylpyridin-2-yl)-4,5-dihydrooxazole(10)2-(benzo[b]thiophen-2-yl)-4,5-dihydrooxazole(10)2-(pyridin-2-ylmethyl)-4,5-dihydrooxazole(14)2-(quinolin-2-yl)-4,5-dihydrooxazole(7)2-phenyl-4,5-dihydrooxazole(18)4,4',5,5'-tetrahydro-2,2'-bioxazole(10)4-(tert-butyl)-2-(pyridin-2-yl)-4,5-dihydrooxazole(16)4-isopropyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(16)bis((3aR,8aS)-3a,8a-dihydro-8H-indeno[1,2-d]oxazol-2-yl)methane(5)bis((3aS,8aR)-3a,8a-dihydro-8H-indeno[1,2-d]oxazol-2-yl)methane(5)bis(4,5-dihydrooxazol-2-yl)methane(22)dihydrooxazole(488)
Piperazines(7422)
(9H-fluoren-9-yl)methyl piperazine-1-carboxylate(7)(R)-2-benzylpiperazine(5)1,4-diphenylpiperazine(7)1-(2-chlorophenyl)piperazine(4)1-(2-fluorophenyl)piperazine(8)1-(3-methoxyphenyl)piperazine(4)1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)piperazine(10)1-(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine(6)1-(methylsulfonyl)piperazine(5)1-(phenylsulfonyl)piperazine(16)1-(piperidin-4-yl)piperazine(6)1-(pyridin-3-yl)piperazine(13)1-(pyridin-3-ylmethyl)piperazine(6)1-benzhydryl-4-benzylpiperazine(1)1-benzhydrylpiperazine(9)1-benzyl 4-(tert-butyl) piperazine-1,4-dicarboxylate(10)1-benzyl-4-methylpiperazine(10)1-ethyl-4-phenylpiperazine(4)1-ethylpiperazine(5)1-isopropylpiperazine(4)1-methyl-4-(pyridin-2-yl)piperazine(5)1-methyl-4-phenylpiperazine(22)1-methylpiperazin-2-one(6)1-methylpiperazine(21)10-(3-(piperazin-1-yl)propyl)-10H-phenothiazine(2)2-(4-(benzyloxy)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(10)2-(piperazin-1-yl)benzonitrile(6)2-(piperazin-1-yl)ethan-1-amine(6)2-(piperazin-1-yl)ethan-1-ol(9)2-(piperazin-1-yl)pyrazine(9)2-(piperazin-1-yl)pyrimidine(11)2-phenylpiperazine(17)3-(piperazin-1-yl)aniline(3)3-(piperazin-1-yl)benzoic acid(3)3-(piperazin-1-yl)pyridazine(7)3-methylpiperazin-2-one(7)4-(piperazin-1-yl)benzaldehyde(5)4-(piperazin-1-yl)pyrimidine(13)benzyl 2-methylpiperazine-1-carboxylate(7)benzyl 3-(hydroxymethyl)piperazine-1-carboxylate(6)benzyl 3-methylpiperazine-1-carboxylate(5)phenyl(piperazin-1-yl)methanone(20)piperazine-2,5-dione(9)tert-butyl 3-oxopiperazine-1-carboxylate(7)tert-butyl 4-(pyridin-2-yl)piperazine-1-carboxylate(12)tert-butyl 4-benzylpiperazine-1-carboxylate(13)tert-butyl 4-phenylpiperazine-1-carboxylate(23)tert-butyl piperazine-1-carboxylate(30)benzothiophene-piperazine(5)benzylpiperazine(14)butylpiperazine(3)cyanopiperazine(3)dimethylpiperazine(40)dioxaborolane-piperazine(40)ethylpiperazine(12)hydroxymethyl-piperazine-carboxylate(17)isobutylpiperazine(6)isopropylpiperazine(9)methylpiperazine(164)oxopiperazine(12)phenylpiperazine(18)piperazin-amine(50)piperazin-pyrimidin(6)piperazine-carboxylate(50)piperazine-carboxylic acid(39)piperazine-hydrochloride(193)piperazine-hydroxyl(73)piperazine-methanol(5)piperazine-pyrazine(15)piperazine-pyridine(145)piperazine-quinoline(49)propylpiperazine(10)pyrazol-piperazine(39)trifluoromethyl-phenyl-piperazin(23)trifluoromethyl-piperazine(46)trimethylpiperazine(5)
Piperidines(21840)
(9H-fluoren-9-yl)methyl piperidine-1-carboxylate(16)(E)-N-(3-oxo-1-phenyl-3-(piperidin-1-yl)prop-1-en-2-yl)benzamide(6)(R)-2-phenylpiperidine(10)(R)-3-aminopiperidin-2-one(3)(R)-N-(piperidin-3-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine(5)(S)-2-phenylpiperidine(11)(S)-N-phenylpiperidine-2-carboxamide(1)1,3-dihydrospiro[indene-2,4'-piperidine](5)1,4'-bipiperidine(8)1-((benzyloxy)carbonyl)piperidine-2-carboxylic acid(5)1-((benzyloxy)carbonyl)piperidine-3-carboxylic acid(3)1-(methylsulfonyl)piperidine(6)1-(phenylsulfonyl)piperidine(23)1-(piperidin-1-yl)ethan-1-one(13)1-(piperidin-4-yl)piperazine(6)1-benzyl 3-methyl piperidine-1,3-dicarboxylate(3)1-benzyl-N-methylpiperidin-3-amine(6)1-benzylpiperidin-3-one(5)1-benzylpiperidin-4-amine(8)1-benzylpiperidin-4-one(8)1-isopropylpiperidine(5)1-methyl-4-phenylpiperidine(4)1-methylpiperidin-2-one(3)1-oxa-8-azaspiro[4.5]decane(5)1-phenylpiperidin-2-one(8)2,2-dimethylpiperidin-4-one(6)2,2-dimethylpiperidine(21)2,7-diazaspiro[3.5]nonane(7)2,8-diazaspiro[4.5]decan-3-one(6)2,8-diazaspiro[4.5]decane(5)2-(piperidin-1-yl)ethan-1-amine(8)2-(piperidin-1-yl)ethan-1-ol(10)2-(piperidin-1-yl)pyridine(11)2-(piperidin-1-yl)pyrimidine(16)2-(piperidin-2-yl)ethan-1-ol(8)2-(piperidin-4-yl)ethan-1-ol(5)2-(piperidin-4-yl)pyridine(5)2-methylpiperidin-4-one(6)2-methylpiperidine(14)2-phenylpiperidine(14)3,3-difluoropiperidin-4-one(3)3,3-difluoropiperidine(29)3,3-dimethylpiperidin-4-one(3)3,3-dimethylpiperidine(5)3-(1-oxoisoindolin-2-yl)piperidine-2,6-dione(21)3-(piperidin-1-yl)pyridine(5)3-(piperidin-4-yl)-1H-indazole(5)3-(piperidin-4-yl)-1H-indole(8)3-(piperidin-4-yl)pyridine(5)3-fluoropiperidin-4-one(3)3-fluoropiperidine(26)3-methoxypiperidine(7)3-methylpiperidin-4-one(3)3-methylpiperidine(10)3-phenylpiperidine(6)4,4-difluoropiperidine(5)4-(1H-pyrazol-1-yl)piperidine(11)4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2,3,6-tetrahydropyridine(11)4-(azetidin-1-yl)piperidine(6)4-(benzyloxy)piperidine(1)4-(piperidin-1-yl)pyrimidine(9)4-(pyrrolidin-1-yl)piperidine(6)4-benzylpiperidine(18)4-hydrazineylpiperidine(5)4-hydroxypiperidin-2-one(3)4-methyl-1-phenylpiperidine(6)4-methylenepiperidine(14)4-oxopiperidine-2-carboxylic acid(3)4-phenylpiperidin-4-ol(10)5-hydroxypiperidin-2-one(3)6-azaspiro[2.5]octane(10)6-oxopiperidine-2-carboxylic acid(3)7-azaspiro[3.5]nonane(7)azepane(25)benzyl 3-hydroxypiperidine-1-carboxylate(7)benzyl 4-oxopiperidine-1-carboxylate(12)benzyl piperidin-4-ylcarbamate(7)benzyl piperidine-1-carboxylate(50)benzyl-3-aminopiperidine-1-carboxylate(6)ethyl piperidine-1-carboxylate(5)ethyl piperidine-2-carboxylate(9)ethyl piperidine-3-carboxylate(13)ethyl piperidine-4-carboxylate(9)methyl 4-oxopiperidine-2-carboxylate(3)methyl piperidine-2-carboxylate(12)methyl piperidine-3-carboxylate(19)N-benzylpiperidin-4-amine(3)N-methylpiperidin-3-amine(5)N-phenylpiperidine-2-carboxamide(2)phenyl(piperidin-1-yl)methanone(21)phenyl(piperidin-4-yl)methanone(21)piperidin-2-ylmethanamine(6)piperidin-2-ylmethanol(11)piperidin-3-amine(17)piperidin-3-ol(27)piperidin-3-one(13)piperidin-3-ylmethanamine(5)piperidin-3-ylmethanol(15)piperidin-4-amine(14)piperidin-4-ol(34)piperidin-4-ylmethanamine(7)piperidin-4-ylmethanol(8)piperidine-2,4-dione(5)piperidine-2,6-dione(13)piperidine-2-carboxylic acid(26)piperidine-3-carboxamide(5)piperidine-3-carboxylic acid(14)piperidine-4-carboxylic acid(12)quinuclidine(12)spiro[benzo[d][1,3]oxazine-4,4'-piperidin]-2(1H)-one(5)spiro[chromane-2,4'-piperidin]-4-one(6)spiro[indene-2,4'-piperidin]-1(3H)-one(5)spiro[indoline-3,4'-piperidin]-2-one(18)spiro[indoline-3,4'-piperidine](5)tert-butyl (2-oxopiperidin-3-yl)carbamate(4)tert-butyl (6-oxopiperidin-3-yl)carbamate(3)tert-butyl (piperidin-3-ylmethyl)carbamate(5)tert-butyl 2-oxopiperidine-1-carboxylate(3)tert-butyl 3-bromopiperidine-1-carboxylate(4)tert-butyl 4-oxopiperidine-1-carboxylate(16)tert-butyl 4-phenoxypiperidine-1-carboxylate(12)tert-butyl 4-phenylpiperidine-1-carboxylate(16)tert-butyl piperidin-3-ylcarbamate(9)acetylpiperidine(3)aminopiperidine(33)benzoylpiperidine(26)benzylpiperidine(32)bipiperidine(12)bromopiperidine(6)carbamoylpiperidine(5)chloropiperidine(6)cyanopiperidine(14)cyclopropylpiperidin(1)difluoropiperidine(19)dimethylpiperidine(46)dioxaborolane-piperidine(4)dioxopiperidine(14)ethylpiperidine(15)ethynylpiperidine(5)fluoropiperidine(33)formylpiperidine(9)hydroxyethyl-piperidine-carboxylate(3)hydroxymethyl-piperidine-carboxylate(23)hydroxypiperidine(46)isopropylpiperidine(11)methoxypiperidine(11)methylenepiperidine(5)methylpiperidine(191)oxoethyl-piperidine-carboxylate(5)oxopiperidine(52)pentamethylpiperidin(1)phenoxypiperidine(59)phenylpiperidine(29)piperidin-amine(1)piperidin-imidazolyl(39)piperidine-benzimidazole(5)piperidine-benzofuran(6)piperidine-carboxylate(530)piperidine-carboxylate-hydrochloride(55)piperidine-carboxylic acid(170)piperidine-ethanol(16)piperidine-hydrochloride(39)piperidine-indole(105)piperidine-pyrimidine(192)piperidine-triazole(20)piperidineacetic(5)piperidinecarboxylate(8)piperidinecarboxylic(6)piperidinemethanol(6)piperidinone(14)piperidinyl-phenyl-boronic acid(36)piperidinyl-phenyl-boronic acid/ester(10)propylpiperidine(8)pyrazole-piperidine(67)pyridin-piperidine(169)tetrahydropyrazolopyridine(8)tetramethylpiperidine(9)trifluoromethyl-phenyl-piperidine(28)trifluoromethyl-piperidine(91)
Purines(2022)
(R)-N-(9-(tetrahydrofuran-2-yl)-9H-purin-6-yl)benzamide(5)2-(acetoxymethyl)-5-(9H-purin-9-yl)tetrahydrofuran-3,4-diyl diacetate(6)2-(hydroxymethyl)-5-(9H-purin-9-yl)tetrahydrofuran-3,4-diol(28)2-chloro-7H-purine(3)2-chloro-9H-purine(5)7H-purin-2-amine(6)7H-purin-6-amine(9)9-((3aS,4R,6aR)-tetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purine(7)9-(tetrahydrofuran-2-yl)-9H-purin-6-amine(30)9-(tetrahydrofuran-2-yl)-9H-purin-6-ol(6)9-(tetrahydrofuran-2-yl)-9H-purine(2)9H-purin-6-amine(3)amino-purin(185)chloro-purine(34)purin-one(52)
Pyrans(1092)
(S)-2-phenylchroman-4-one(17)1-(tetrahydro-2H-pyran-2-yl)-1H-indazole(17)2,2-dimethylchromane(5)2-(2-oxo-2H-chromen-4-yl)acetic acid(4)2-oxo-2H-chromene-3-carbonitrile(5)2-oxo-2H-chromene-3-carboxylic acid(14)2H-chromene(16)3,4-dihydro-2H-pyran(11)3,6-dihydro-2H-pyran(8)3-acetyl-2H-chromen-2-one(3)3-hydroxy-2-phenyl-4H-chromen-4-one(20)4-(trifluoromethyl)-2H-chromen-2-one(6)4-hydroxy-2H-chromen-2-one(10)4-methyl-2H-chromen-2-one(26)5-fluorochroman-4-one(3)5-fluorochromane(6)5-hydroxy-2-phenyl-4H-chromen-4-one(36)5-methoxy-2-phenyl-4H-chromen-4-one(10)6-bromochroman-4-one(6)6-chlorochroman-4-one(6)6-chlorochromane(6)6-fluorochroman-4-one(5)6-fluorochromane(6)6-methoxychromane(5)7-(diethylamino)-2H-chromen-2-one(8)7-bromochroman-4-one(4)7-bromochromane(7)7-chlorochromane(5)7-fluorochroman-4-one(5)7-hydroxy-2H-chromen-2-one(21)7-methoxy-2H-chromen-2-one(11)7-methoxychroman-4-one(3)8-bromochroman-4-one(4)8-bromochromane(6)8-chlorochromane(5)8-fluorochromane(5)8-methylchroman-4-one(4)8-methylchromane(10)chroman-2-one(5)chroman-2-ylmethanamine(3)chroman-3-amine(3)chroman-4-amine(20)chroman-4-ol(4)chromane-2-carboxylic acid(6)chromane-3-carboxylic acid(4)dihydro-2H-pyran-2,6(3H)-dione(5)ethyl 2-oxo-2H-chromene-3-carboxylate(5)isochromane(11)spiro[chromane-2,4'-piperidin]-4-one(6)tetrahydro-2H-pyran-2-one(16)tetrahydro-2H-thiopyran(10)tetrahydro-4H-pyran-4-one(12)thiochroman-4-one(10)amino-chromen(50)benzopyran(7)benzopyran-carboxylate(3)benzopyran-ethylamine(1)benzopyran-nitrile(1)bromo-chromen(36)chlorochroman(11)chroman-carboxylate(4)chroman-carboxylic(26)chroman-one(115)chromen-one(38)chromene-carbonitrile(4)chromene-carboxylic acid(27)furochromen(7)hydroxychroman-one(5)methoxy-chromen-one(6)methoxy-pyran(144)methyl-chromen(4)methyl-pyran(6)phenyl-chromen-one(154)phenylchroman(4)pyran-carboxylic acid(60)pyran-one(165)pyranopyridine(4)pyranoquinoline(5)pyranyl-carboxylate(7)thioglucopyranoside(4)trifluoromethyl-chroman(3)
Pyrazines(3350)
1,2,3,4-tetrahydropyrido[2,3-b]pyrazine(5)1-(pyrazin-2-yl)ethan-1-one(5)1H-pyrazolo[3,4-b]pyrazine(5)2,3-diphenylpyrazine(5)2,5-dibromopyrazine(4)2,5-dichloropyrazine(4)2,6-dibromopyrazine(9)2,6-dichloropyrazine(14)2-(piperazin-1-yl)pyrazine(9)2-(trifluoromethyl)pyrazine(6)2-bromo-3-methoxypyrazine(5)2-bromo-3-methylpyrazine(5)2-bromo-5-chloropyrazine(6)2-bromo-6-chloropyrazine(5)2-bromo-6-methylpyrazine(5)2-bromopyrazine(15)2-chloro-3-methylpyrazine(7)2-chloro-6-methylpyrazine(12)2-chloropyrazine(24)2-ethoxypyrazine(3)2-ethylpyrazine(9)2-fluoropyrazine(7)2-iodopyrazine(25)2-methoxypyrazine(27)2-methylpyrazine(16)2-phenylpyrazine(12)3-chloropyrazin-2-amine(6)3-chloropyrazine-2-carbonitrile(6)4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine(7)4-(pyrazin-2-yl)morpholine(5)5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine(11)5,6,7,8-tetrahydroimidazo[1,2-a]pyrazine(15)5,6,7,8-tetrahydroimidazo[1,5-a]pyrazine(11)5-bromopyrazin-2-amine(13)5-bromopyrazin-2-ol(5)5-chloropyrazin-2-amine(9)5H-pyrrolo[2,3-b]pyrazine(14)6-bromopyrazin-2-amine(5)6-chloropyrazin-2-amine(11)[1,2,4]triazolo[1,5-a]pyrazine(9)[1,2,4]triazolo[4,3-a]pyrazine(11)ethyl pyrazine-2-carboxylate(10)imidazo[1,5-a]pyrazine(9)methyl 3-bromopyrazine-2-carboxylate(5)methyl 6-bromopyrazine-2-carboxylate(7)methyl 6-chloropyrazine-2-carboxylate(5)methyl pyrazine-2-carboxylate(19)pyrazin-2-amine(22)pyrazin-2-ol(7)pyrazin-2-ylmethanamine(11)Pyrazine Derivatives(99)pyrazine-2-carbaldehyde(15)pyrazine-2-carbohydrazide(3)pyrazine-2-carbonitrile(15)pyrazine-2-carboxamide(10)pyrazine-2-carboxylic acid(34)pyrazolo[1,5-a]pyrazine(5)pyrido[2,3-b]pyrazine(12)pyrido[3,4-b]pyrazine(11)tert-butyl pyrazin-2-ylcarbamate(8)aminopyrazine(11)bromopyrazine(25)chloro-triazolopyridazine(3)chloropyrazine(43)cyanopyrazine(3)dibromopyrazine(7)dichloropyrazine(21)dihydropyrazine(6)dimethylpyrazine(30)ethoxypyrazine(5)ethylpyrazine(7)fluoropyrazine(6)hydrazinylpyrazine(4)hydroxypyrazine(4)imidazopyrazine(99)iodopyrazine(17)methoxypyrazine(36)methylpyrazine(73)octahydropyrazino(3)oxazolopyrazin-one-hydrochloride(3)phenylpyrazin(5)piperazine-pyrazine(15)pyrazin-amine(89)pyrazin-one(60)pyrazine-nitrile(43)pyrazinecarboxylate(95)pyrazinecarboxylic(69)pyrazino(5)pyrazino-nitrile(23)pyrazolopyrazine(34)pyridopyrazine(39)pyrrolopyrazine(47)triazolopyrazine(49)trifluoromethyl-phenyl-pyrazin(1)trifluoromethyl-pyrazine(39)trimethylpyrazine(2)
Pyrazoles(16093)
(1-methyl-1H-pyrazol-3-yl)methanol(7)(1-phenyl-1H-pyrazol-4-yl)boronic acid(4)1,3-dimethyl-1H-pyrazole(28)1,3-diphenyl-1H-pyrazole(6)1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole(6)1,5-diphenyl-1H-pyrazole(13)1-(2-chlorophenyl)-1H-pyrazole(3)1-(3-chlorophenyl)-1H-pyrazole(3)1-(4-methoxybenzyl)-1H-pyrazole(5)1-(azetidin-3-yl)-1H-pyrazole(7)1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole(9)1-(tetrahydro-2H-pyran-4-yl)-1H-pyrazole(6)1-benzyl-1H-pyrazolo[3,4-b]pyridine(9)1-benzyl-4-bromo-1H-pyrazole(5)1-benzyl-4-iodo-1H-pyrazole(5)1-cyclopentyl-1H-pyrazole(5)1-cyclopropyl-1H-pyrazole(9)1-methyl-1H-pyrazol-3-amine(14)1-methyl-3-phenyl-1H-pyrazole(21)1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole(6)1-methyl-5-phenyl-1H-pyrazole(7)1-phenyl-1H-pyrazol-3-amine(3)1-phenyl-1H-pyrazol-3-ol(3)1-phenyl-1H-pyrazol-5-amine(4)1-phenyl-1H-pyrazole-3-carboxylic acid(9)1-phenyl-1H-pyrazole-4-carbaldehyde(4)1-phenyl-1H-pyrazole-4-carbonitrile(13)1-phenyl-1H-pyrazole-4-carboxylic acid(17)1-phenyl-1H-pyrazole-5-carboxylic acid(3)1-phenyl-3-(trifluoromethyl)-1H-pyrazole(5)1-trityl-1H-pyrazole(5)1H-pyrazol-4-amine(6)1H-pyrazol-4-ol(3)1H-pyrazol-5-amine(19)1H-pyrazole-4-carbaldehyde(17)1H-pyrazole-4-carbonitrile(5)1H-pyrazole-4-carboxylic acid(6)1H-pyrazole-4-sulfonyl chloride(8)1H-pyrazole-5-carboxamide(3)1H-pyrazole-5-carboxylic acid(10)1H-pyrazolo[3,4-b]pyrazine(5)1H-pyrazolo[3,4-b]pyridin-5-amine(3)1H-pyrazolo[3,4-d]pyrimidin-4-amine(5)1H-pyrazolo[4,3-d]pyrimidine(5)2,3-dihydropyrazolo[5,1-b]oxazole(7)2,4-dihydro-3H-pyrazol-3-one(10)2,6-di(1H-pyrazol-1-yl)pyridine(5)2-(1H-pyrazol-1-yl)pyridine(16)2-(1H-pyrazol-1-yl)pyrimidine(6)2-(1H-pyrazol-3-yl)pyridine(5)2-phenyl-2,4-dihydro-3H-pyrazol-3-one(23)3-(tert-butyl)-1-phenyl-1H-pyrazole(4)3-bromo-1-methyl-1H-pyrazole(16)3-bromopyrazolo[1,5-a]pyridine(6)3-bromopyrazolo[1,5-a]pyrimidine(11)3-chloro-1-methyl-1H-pyrazole(7)3-cyclopropyl-1H-pyrazole(5)3-iodo-1-methyl-1H-pyrazole(6)3-iodo-1H-pyrazolo[3,4-b]pyridine(4)3-methyl-1-phenyl-1H-pyrazole(23)3-phenyl-1H-pyrazol-5-amine(11)4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine(7)4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine(14)4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine(7)4,5,6,7-tetrahydropyrazolo[1,5-a]pyridine(6)4-(1H-pyrazol-1-yl)benzoic acid(3)4-(1H-pyrazol-1-yl)piperidine(11)4-bromo-1-methyl-1H-pyrazole(14)4-bromo-1-phenyl-1H-pyrazole(11)4-bromo-1H-pyrazolo[3,4-b]pyridine(4)4-bromopyrazolo[1,5-a]pyridine(8)4-chloro-1H-pyrazole(10)4-chloro-1H-pyrazolo[3,4-b]pyridine(5)4-chloro-1H-pyrazolo[3,4-d]pyrimidine(5)4-iodo-1H-pyrazole(9)4-methyl-1-phenyl-1H-pyrazole(6)4-methyl-1H-pyrazole(11)4-nitro-1H-pyrazole(8)4-phenyl-1H-pyrazole(18)5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole(8)5-(difluoromethyl)-1H-pyrazole(6)5-bromo-1H-pyrazole(11)5-bromo-1H-pyrazolo[3,4-b]pyridine(11)5-bromo-1H-pyrazolo[3,4-c]pyridine(5)5-chloro-1H-pyrazole(8)5-chloro-1H-pyrazolo[3,4-b]pyridine(3)5-chloropyrazolo[1,5-a]pyrimidine(24)5-cyclopropyl-1H-pyrazole(6)5-iodo-1H-pyrazole(4)5-nitro-1H-pyrazole(9)5-nitro-1H-pyrazolo[3,4-b]pyridine(3)5-phenyl-1H-pyrazole-3-carboxylic acid(12)6,7-dihydro-5H-pyrazolo[5,1-b][1,3]oxazine(13)6-bromopyrazolo[1,5-a]pyridine(8)6-bromopyrazolo[1,5-a]pyrimidine(7)6-chloro-1H-pyrazolo[3,4-d]pyrimidine(12)7-chloropyrazolo[1,5-a]pyrimidine(8)ethyl 1-phenyl-1H-pyrazole-3-carboxylate(4)ethyl 1-phenyl-1H-pyrazole-4-carboxylate(13)ethyl 1H-pyrazole-4-carboxylate(11)ethyl pyrazolo[1,5-a]pyridine-3-carboxylate(11)ethyl pyrazolo[1,5-a]pyrimidine-3-carboxylate(6)methyl 1H-pyrazole-4-carboxylate(9)methyl 1H-pyrazolo[3,4-b]pyridine-5-carboxylate(3)methyl pyrazolo[1,5-a]pyridine-3-carboxylate(16)pyrazolo[1,5-a]pyrazine(5)pyrazolo[1,5-a]pyridine-2-carboxylic acid(5)pyrazolo[1,5-a]pyridine-3-carboxylic acid(12)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid(7)tert-butyl 1H-pyrazolo[3,4-b]pyridine-1-carboxylate(7)tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole-1-carboxylate(3)amino-pyrazole-carboxylate(66)amino-pyrazolopyridine(13)aminopyrazolo(5)benzylpyrazol-amide(4)bipyrazole(5)bromo-methyl-pyrazole(169)bromo-pyrazol-amide(7)bromo-pyrazole-carboxylate(60)bromo-pyrazolopyridine(117)bromo-pyrazolopyrimidine(44)bromo-trifluoromethyl-pyrazole(7)bromopyrazolo(34)chloro-methyl-pyrazole(155)chloro-pyrazolamide(1)chloro-pyrazole(23)chloro-pyrazole-carboxylate(36)chloro-pyrazolopyridine(38)chloro-pyrazolopyrimidine(76)chloropyrazolo(33)dichloropyrazolo(11)difluoromethyl-pyrazole(30)difluoromethyl-pyrazole-carboxylate(5)dihydropyrazolo(28)dimethyl-pyrazol-amide(38)dimethyl-pyrazole-carboxylate(16)dimethyl-pyrazole-carboxylic acid(13)dimethylpyrazole(5)dimethylpyrazolo(6)dioxaborolan-pyrazole(161)ethyl-pyrazol-amide(4)ethylpyrazole(7)fluoropyrazolo(9)hydroxy-pyrazole(74)hydroxy-pyrazolopyrimidine(6)hydroxypyrazolo(10)iodo-methyl-pyrazole(36)iodo-pyrazole-carboxylate(19)iodopyrazolo(4)isopropyl-pyrazol-amide(3)methoxy-pyrazolo-pyridine(30)methoxypyrazolo(16)methy-pyrazolooxazine(4)methyl-hydrox-ypyrazole(6)methyl-nitro-pyrazole(49)methyl-pyrazol-one(81)methyl-pyrazole-carboxylic acid(147)methyl-pyrazolone(54)methyl-pyrazolopyridine(143)methylpyrazole(7)methylpyrazolo(32)nitro-pyrazole-carboxylate(6)phenyl-pyrazol-amide(83)phenyl-pyrazole-carboxylate(39)phenylpyrazole(3)pyrazo-morpholine(16)pyrazol-amine(43)pyrazol-boronic acid(66)pyrazol-methanol(34)pyrazol-one(39)pyrazol-piperazine(39)pyrazol-pyridine(513)pyrazole-carbonitrile(37)pyrazole-carboxylate(40)pyrazole-carboxylic acid(280)pyrazole-ethanol(11)pyrazole-hydrochloride(78)pyrazole-methamine(3)pyrazole-phenyl-boronic-acid(3)pyrazole-piperidine(67)pyrazolo-pyrimidine(261)pyrazolooxazine(17)pyrazolopyrazine(34)pyrazolopyridin-ol(8)pyrazolopyridin-one(24)pyrazolopyridine(7)pyrazolopyridine-carbonitrile(16)pyrazolopyridine-carboxylate(63)pyrazolopyridine-carboxylic acid(48)pyrazolopyrimid-one(28)pyrazolopyrimidine(261)pyrazolotriazine(4)pyridine-pyrazole(513)pyrrolopyrazole(51)tetrahydropyran-pyrazole(6)tetrahydropyranopyrazole(4)tetrahydropyrazolo(23)trifluoromethyl-pyrazol-amide(24)trifluoromethyl-pyrazole(36)trifluoromethyl-pyrazolone(3)
Pyridazines(2753)
2-methylpyridazin-3(2H)-one(7)3,6-dichloropyridazine(20)3-(piperazin-1-yl)pyridazine(7)3-(trifluoromethyl)pyridazine(3)3-bromopyridazine(25)3-chloro-4-methylpyridazine(7)3-chloropyridazine(44)3-iodopyridazine(5)3-methoxypyridazine(14)3-methylpyridazine(6)3-phenylpyridazine(8)4-aminopyridazin-3(2H)-one(4)4-bromopyridazin-3(2H)-one(3)4-bromopyridazine(5)4-chloropyridazin-3(2H)-one(4)4-chloropyridazine(16)4-methoxypyridazine(5)4-methylpyridazine(3)6-chloropyridazin-3(2H)-one(5)6-chloropyridazin-3-amine(5)[1,2,4]triazolo[4,3-b]pyridazine(10)ethyl pyridazine-3-carboxylate(6)methyl pyridazine-3-carboxylate(7)pyridazin-3-amine(18)pyridazin-3-ol(3)pyridazin-4-amine(10)pyridazine-3-carboxylic acid(7)aminopyridazine(12)bromopyridazine(9)chloropyridazine(33)dichloropyridazine(17)dihydropyridazine(26)dimethylpyridazine(9)ethoxypyridazine(4)ethylpyridazin(2)hexahydropyridazine(9)Hexahydropyridazines(6)hydroxypyridazine(5)imidazopyridazine(62)methoxypyridazine(17)methylpyridazine(31)phenylpyridazin(12)pyridazin-amine(55)pyridazin-one(78)pyridazine-carboxylate(58)pyridazinecarboxylic(36)pyrrolopyridazine(3)triazolopyridazine(12)trifluoromethyl-pyridazine(23)
Pyridines(70453)
(2-chloropyridin-4-yl)boronic acid(6)(3-bromopyridin-2-yl)methanol(7)(3-chloropyridin-2-yl)methanamine(6)(3-fluoropyridin-2-yl)methanamine(5)(3-methylpyridin-2-yl)methanamine(5)(3-methylpyridin-2-yl)methanol(10)(3aR,8aS)-2-(pyridin-2-yl)-3a,8a-dihydro-8H-indeno[1,2-d]oxazole(6)(3aS,8aR)-2-(pyridin-2-yl)-3a,8a-dihydro-8H-indeno[1,2-d]oxazole(7)(4R,5S)-4,5-diphenyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(5)(5-(trifluoromethyl)pyridin-3-yl)boronic acid(4)(5-bromopyridin-2-yl)methanamine(5)(5-bromopyridin-2-yl)methanol(5)(5-chloropyridin-2-yl)methanamine(5)(6-methylpyridin-2-yl)methanamine(6)(6-methylpyridin-2-yl)methanol(5)(6-phenoxypyridin-3-yl)boronic acid(5)(R)-3-(pyrrolidin-2-yl)pyridine(9)(R)-4-benzyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(7)(R)-4-phenyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(7)(S)-4-benzyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(8)(S)-4-phenyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(6)1,1'-diphenyl-[4,4'-bipyridine]-1,1'-diium(7)1,10-phenanthrolin-4-ol(3)1,2,3,4-tetrahydropyrido[2,3-b]pyrazine(5)1,2,3,6-tetrahydropyridine(21)1,3-dihydro-2H-pyrrolo[2,3-b]pyridin-2-one(19)1,3-dihydro-2H-pyrrolo[2,3-c]pyridin-2-one(5)1,3-dihydro-2H-pyrrolo[3,2-b]pyridin-2-one(11)1,4-di(pyridin-4-yl)benzene(5)1-(5-bromopyridin-2-yl)piperazine(6)1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridine(30)1-(pyridin-2-yl)ethan-1-amine(15)1-(pyridin-2-yl)ethan-1-one(36)1-(pyridin-2-yl-l2-azaneyl)ethan-1-one(12)1-(pyridin-3-yl)ethan-1-amine(17)1-(pyridin-3-yl)ethan-1-one(29)1-(pyridin-3-yl)piperazine(13)1-(pyridin-3-ylmethyl)piperazine(6)1-(pyridin-4-yl)ethan-1-amine(23)1-(pyridin-4-yl)ethan-1-one(14)1-benzyl-1,2,3,6-tetrahydropyridine(9)1-benzyl-1H-pyrazolo[3,4-b]pyridine(9)1-methyl-4-(pyridin-2-yl)piperazine(5)1-methylpyridin-1-ium iodide(5)1-phenyl-3-(4-(pyridin-4-yloxy)phenyl)urea(8)1H-imidazo[4,5-b]pyridine(14)1H-imidazo[4,5-c]pyridine(12)1H-pyrazolo[3,4-b]pyridin-5-amine(3)1H-pyrrolo[2,3-c]pyridin-7-ol(3)1H-pyrrolo[2,3-c]pyridine-2-carboxylic acid(4)1H-pyrrolo[3,2-b]pyridine-3-carboxylic acid(4)2,2':6',2''-terpyridine(15)2,2-dimethyl-1-(pyridin-2-yl-l2-azaneyl)propan-1-one(7)2,3'-bipyridine(11)2,3,4,5-tetrahydro-1H-pyrido[4,3-b]indole(12)2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole(13)2,3,5-trichloropyridine(13)2,3,6-trichloropyridine(16)2,3-dibromopyridine(21)2,3-dichloropyridine(38)2,3-difluoropyridine(13)2,3-dihydro-1H-pyrrolo[2,3-b]pyridine(10)2,3-dihydro-1H-pyrrolo[2,3-c]pyridine(5)2,3-dihydro-1H-pyrrolo[3,2-c]pyridine(7)2,3-dihydro-1H-pyrrolo[3,4-c]pyridine(6)2,3-dihydro-[1,4]dioxino[2,3-b]pyridine(5)2,3-dihydropyridin-2-one(5)2,3-dimethoxypyridine(7)2,3-dimethylpyridine(8)2,4,6-trichloropyridine(5)2,4,6-triphenylpyridine(7)2,4-dibromopyridine(5)2,4-dichloro-6-methylpyridine(8)2,4-dichloropyridine(50)2,4-dimethylpyridine(5)2,5-dibromopyridine(26)2,5-dichloropyridine(32)2,5-difluoropyridine(8)2,6-bis(4,5-dihydrooxazol-2-yl)pyridine(12)2,6-di(1H-pyrazol-1-yl)pyridine(5)2,6-dibromo-3-fluoropyridine(3)2,6-dibromopyridin-4-amine(3)2,6-dibromopyridine(28)2,6-dichloro-3-fluoropyridine(10)2,6-dichloro-3-methylpyridine(5)2,6-dichloro-3-nitropyridine(6)2,6-dichloro-4-iodopyridine(4)2,6-dichloro-4-methylpyridine(5)2,6-dichloronicotinonitrile(8)2,6-dichloropyridin-3-amine(11)2,6-dichloropyridine(49)2,6-difluoropyridine(29)2,6-dimethoxypyridine(11)2,6-dimethylpyridin-4-ol(4)2,6-dimethylpyridine(26)2-(1H-pyrazol-1-yl)pyridine(16)2-(1H-pyrazol-3-yl)pyridine(5)2-(2,2,2-trifluoroethoxy)pyridine(8)2-(2-chlorophenyl)pyridine(3)2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(6)2-(4-fluorophenyl)pyridine(12)2-(6-benzhydrylpyridin-2-yl)-4,5-dihydrooxazole(10)2-(6-cyclopropylpyridin-2-yl)-4,5-dihydrooxazole(10)2-(benzyloxy)pyridine(18)2-(bromomethyl)pyridine(27)2-(chloromethyl)-3-methylpyridine(12)2-(chloromethyl)pyridine(12)2-(chloromethyl)pyridine 1-oxide(4)2-(difluoromethyl)pyridine(21)2-(methylsulfonyl)pyridine(11)2-(methylthio)pyridine(5)2-(p-tolyl)pyridine(6)2-(phenoxymethyl)pyridine(5)2-(piperidin-1-yl)pyridine(11)2-(piperidin-4-yl)pyridine(5)2-(pyridin-2-yl)-1H-benzo[d]imidazole(5)2-(pyridin-2-yl)acetonitrile(8)2-(pyridin-2-yl)pyrimidine(8)2-(pyridin-2-ylmethyl)-4,5-dihydrooxazole(14)2-(pyridin-3-yl)acetic acid(7)2-(pyridin-3-yl)acetonitrile(6)2-(pyrrolidin-2-yl)pyridine(5)2-(thiophen-2-yl)pyridine(5)2-(trifluoromethoxy)pyridine(6)2-(trifluoromethyl)pyridine(6)2-aminonicotinonitrile(6)2-benzylpyridine(12)2-bromo-3-(trifluoromethyl)pyridine(4)2-bromo-3-chloropyridine(25)2-bromo-3-fluoropyridine(26)2-bromo-3-iodopyridine(6)2-bromo-3-methoxypyridine(7)2-bromo-3-methylpyridine(25)2-bromo-3-nitropyridine(5)2-bromo-4-chloropyridine(7)2-bromo-4-methylpyridine(6)2-bromo-5-(trifluoromethyl)pyridine(6)2-bromo-5-chloropyridine(22)2-bromo-5-fluoropyridine(33)2-bromo-5-methoxypyridine(11)2-bromo-5-methylpyridine(19)2-bromo-5-nitropyridine(11)2-bromo-6-(trifluoromethyl)pyridine(6)2-bromo-6-chloropyridine(23)2-bromo-6-fluoropyridine(21)2-bromo-6-iodopyridine(5)2-bromo-6-methoxypyridine(15)2-bromo-6-methylpyridine(17)2-bromo-6-phenylpyridine(4)2-bromonicotinic acid(5)2-bromopyridin-3-amine(10)2-bromopyridin-3-ol(7)2-bromopyridine(10)2-bromopyridine 1-oxide(6)2-chloro-3,6-dimethylpyridine(4)2-chloro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(4)2-chloro-3-(trifluoromethyl)pyridine(12)2-chloro-3-fluoropyridin-4-amine(3)2-chloro-3-fluoropyridine(20)2-chloro-3-iodopyridin-4-amine(3)2-chloro-3-iodopyridine(13)2-chloro-3-methoxypyridine(12)2-chloro-3-methylpyridine(17)2-chloro-3-nitropyridine(10)2-chloro-4,6-dimethylpyridine(7)2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(14)2-chloro-4-(trifluoromethyl)pyridine(9)2-chloro-4-ethoxypyridine(5)2-chloro-4-fluoropyridine(10)2-chloro-4-iodopyridine(19)2-chloro-4-methoxypyridine(22)2-chloro-4-methylpyridine(31)2-chloro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(6)2-chloro-5-(trifluoromethyl)pyridine(4)2-chloro-5-fluoropyridin-4-amine(3)2-chloro-5-fluoropyridine(16)2-chloro-5-iodopyridine(18)2-chloro-5-methylpyridine(11)2-chloro-5-nitropyridine(8)2-chloro-6-(trifluoromethyl)pyridine(16)2-chloro-6-fluoropyridine(14)2-chloro-6-iodopyridine(9)2-chloro-6-methoxypyridine(24)2-chloro-6-methyl-3-nitropyridine(4)2-chloro-6-methylnicotinonitrile(10)2-chloro-6-methylpyridine(49)2-chloroisonicotinaldehyde(14)2-chloroisonicotinic acid(17)2-chloroisonicotinonitrile(7)2-chloronicotinic acid(7)2-chloronicotinonitrile(9)2-chloropyridin-3-amine(11)2-chloropyridin-3-ol(9)2-chloropyridin-4-amine(28)2-chloropyridine 1-oxide(10)2-cyclopropyl-4-phenylquinoline(12)2-cyclopropylpyridine(20)2-ethoxypyridine(20)2-ethylpyridine(9)2-ethynylpyridine(29)2-fluoro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(7)2-fluoro-3-iodopyridine(8)2-fluoro-3-methylpyridine(14)2-fluoro-4-methylpyridine(6)2-fluoro-5-methoxypyridine(4)2-fluoro-5-methylpyridine(6)2-fluoro-6-methoxypyridine(6)2-fluoro-6-methylpyridine(20)2-fluoropyridine(15)2-hydrazineylpyridine(32)2-iodo-6-methylpyridine(5)2-iodopyridine(27)2-isopropoxypyridine(16)2-isopropylpyridine(14)2-methoxy-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(7)2-methoxy-3-(trifluoromethyl)pyridine(6)2-methoxy-4-methylpyridine(5)2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(5)2-methoxy-5-(trifluoromethyl)pyridine(4)2-methoxy-5-methylpyridine(5)2-methoxy-5-nitropyridine(8)2-methoxy-6-methylpyridine(14)2-methoxyisonicotinic acid(4)2-methoxypyridin-3-amine(6)2-methoxypyridine(197)2-methyl-1,10-phenanthroline(3)2-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(5)2-methyl-3-nitropyridine(9)2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(3)2-methylimidazo[1,2-a]pyridine(8)2-methylpyridin-3-ol(9)2-methylpyridin-4-amine(7)2-methylpyridine 1-oxide(9)2-nitropyridine(40)2-phenoxy-5-(trifluoromethyl)pyridine(7)2-phenylimidazo[1,2-a]pyridine(12)2-propoxypyridine(9)2H-pyrido[3,2-b][1,4]oxazin-3(4H)-one(8)3,3'-bipyridine(7)3,4-dihydro-2H-pyrido[3,2-b][1,4]oxazine(5)3,5-dibromopyridine(12)3,5-dichloropyridine(16)3,5-difluoropyridine(12)3,5-dimethoxypyridine(4)3,5-dimethylpyridine(8)3,5-diphenylpyridine(5)3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5-(trifluoromethyl)pyridine(3)3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(74)3-(benzyloxy)pyridine(19)3-(bromomethyl)pyridine(18)3-(chloromethyl)pyridine(22)3-(cyclopropylmethyl)pyridine(6)3-(difluoromethyl)pyridine(5)3-(methylsulfonyl)pyridine(5)3-(piperidin-1-yl)pyridine(5)3-(piperidin-4-yl)pyridine(5)3-(pyrrolidin-2-yl)pyridine(6)3-(tert-butyl)pyridine(5)3-(trifluoromethyl)-1H-pyrrolo[2,3-b]pyridine(3)3-(trifluoromethyl)pyridin-2-ol(3)3-(trifluoromethyl)pyridine(52)3-bromo-1H-pyrrolo[3,2-b]pyridine(6)3-bromo-2-chloropyridin-4-amine(3)3-bromo-2-chloropyridine(20)3-bromo-2-fluoropyridine(21)3-bromo-2-methoxypyridine(20)3-bromo-2-methylpyridine(27)3-bromo-4-methylpyridine(13)3-bromo-5-chloropyridine(14)3-bromo-5-fluoropyridine(18)3-bromo-5-methylpyridine(14)3-bromo-5-nitropyridine(10)3-bromopicolinic acid(9)3-bromopicolinonitrile(13)3-bromopyrazolo[1,5-a]pyridine(6)3-bromopyridin-2-amine(30)3-bromopyridin-2-ol(14)3-bromopyridine 1-oxide(8)3-chloro-2-fluoropyridine(13)3-chloro-2-methoxypyridine(16)3-chloro-2-methylpyridine(4)3-chloro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(5)3-chloro-5-fluoropyridine(10)3-chloro-5-methylpyridine(7)3-chloro-5-nitropyridine(5)3-chloroimidazo[1,2-a]pyridine(3)3-chloropicolinic acid(9)3-chloropicolinonitrile(6)3-chloropyridin-2-amine(17)3-chloropyridin-2-ol(9)3-cyclopropylpyridine(28)3-ethoxypyridine(12)3-ethylpyridine(11)3-ethynylpyridine(19)3-fluoro-2-methoxypyridine(14)3-fluoro-2-methylpyridine(15)3-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(7)3-fluoropicolinic acid(6)3-fluoropicolinonitrile(5)3-fluoropyridin-2-amine(12)3-fluoropyridin-4-amine(4)3-hydrazineylpyridine(8)3-iodo-1H-pyrazolo[3,4-b]pyridine(4)3-iodo-1H-pyrrolo[3,2-b]pyridine(6)3-iodo-1H-pyrrolo[3,2-c]pyridine(4)3-iodopyridin-2-amine(12)3-iodopyridin-2-ol(4)3-iodopyridin-4-amine(6)3-iodopyridin-4-ol(4)3-iodopyridine(37)3-methoxypyridin-2-amine(5)3-methoxypyridine(52)3-methyl-1H-pyrrolo[2,3-b]pyridine(5)3-methyl-2-phenylpyridine(3)3-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(6)3-methyl-5-nitropyridine(4)3-methylimidazo[1,2-a]pyridine(3)3-methylpicolinic acid(9)3-methylpyridin-2-amine(7)3-methylpyridine(50)3-methylpyridine 1-oxide(7)3-nitropyridin-2-ol(7)3-nitropyridin-4-amine(5)3-nitropyridine(54)3-phenoxypyridine(12)4'-phenyl-2,2':6',2''-terpyridine(13)4,4'-bipyridine(10)4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine(7)4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine(14)4,5,6,7-tetrahydro-3H-imidazo[4,5-c]pyridine(5)4,5,6,7-tetrahydrothiazolo[5,4-c]pyridine(10)4,5,6,7-tetrahydrothieno[2,3-c]pyridine(10)4,5,6,7-tetrahydrothieno[3,2-c]pyridine(9)4,6-dichloropyridin-2-amine(6)4,6-dichloropyridin-3-amine(4)4,7-diphenyl-1,10-phenanthroline(5)4-(2,4-diphenyl-1H-imidazol-5-yl)pyridine(5)4-(benzyloxy)pyridine(8)4-(bromomethyl)pyridine(8)4-(pyridin-2-yl)morpholine(21)4-(pyridin-3-yl)pyrimidine(5)4-(pyridin-4-yl)morpholine(5)4-(tert-butyl)-2-(pyridin-2-yl)-4,5-dihydrooxazole(16)4-(tert-butyl)pyridine(5)4-(trifluoromethyl)nicotinic acid(3)4-(trifluoromethyl)pyridine(14)4-bromo-2-chloropyridine(29)4-bromo-2-fluoropyridine(4)4-bromo-2-methoxypyridine(4)4-bromo-3-methylpyridine(5)4-bromopyrazolo[1,5-a]pyridine(8)4-bromopyridine(31)4-chloro-1H-pyrazolo[4,3-c]pyridine(7)4-chloro-1H-pyrrolo[2,3-b]pyridine(13)4-chloro-1H-pyrrolo[3,2-c]pyridine(10)4-chloro-2,6-dimethylpyridine(4)4-chloro-2-methoxypyridine(6)4-chloro-3-methylpyridine(7)4-chloronicotinic acid(4)4-chloropyridin-2-amine(6)4-chloropyridine(31)4-cyclopropylpyridine(7)4-ethoxypyridine(7)4-ethynylpyridine(8)4-iodopyridine(17)4-isopropyl-2-(pyridin-2-yl)-4,5-dihydrooxazole(16)4-methoxypyridine(67)4-methyl-2,2'-bipyridine(8)4-methyl-2-phenylpyridine(3)4-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(5)4-methyl-3-nitropyridine(8)4-methylnicotinic acid(5)4-methylpyridine(24)4-nitropyridine(11)4-phenoxypyridine(7)4-phenyl-1,2,3,6-tetrahydropyridine(7)4-phenyl-1,4-dihydropyridine(10)5,6,7,8-tetrahydroimidazo[1,2-a]pyridine(12)5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine(7)5,6,7,8-tetrahydropyrido[4,3-d]pyrimidine(17)5,6-dichloropyridin-2-amine(4)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)nicotinonitrile(4)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-amine(5)5-(trifluoromethyl)pyridin-2-amine(5)5-(trifluoromethyl)pyridin-2-ol(6)5-benzyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine(7)5-bromo-2-chloro-4-methylpyridine(5)5-bromo-2-chloropyridine(40)5-bromo-2-fluoropyridine(22)5-bromo-2-iodopyridine(5)5-bromo-2-methoxypyridine(29)5-bromo-2-methylpyridine(23)5-bromo-2-phenylpyridine(3)5-bromo-4-methylpyridin-2-amine(5)5-bromo-6-chloropyridin-2-amine(3)5-bromonicotinaldehyde(7)5-bromonicotinic acid(6)5-bromonicotinonitrile(6)5-bromopicolinic acid(11)5-bromopicolinonitrile(13)5-bromopyridin-2-amine(35)5-bromopyridin-2-ol(10)5-bromopyridin-3-amine(10)5-bromopyridin-3-ol(6)5-chloro-1H-pyrazolo[3,4-b]pyridine(3)5-chloro-1H-pyrazolo[4,3-b]pyridine(6)5-chloro-1H-pyrrolo[2,3-c]pyridine(5)5-chloro-1H-pyrrolo[3,2-b]pyridine(6)5-chloro-2-fluoropyridine(13)5-chloro-2-methoxypyridine(10)5-chloro-2-methylpyridine(10)5-chloroimidazo[1,2-a]pyridine(3)5-chloropicolinic acid(7)5-chloropicolinonitrile(6)5-chloropyridin-2-amine(26)5-chloropyridin-2-ol(12)5-chloropyridin-3-amine(7)5-fluoro-2-methoxypyridine(15)5-fluoro-2-methylpyridine(8)5-fluoropicolinonitrile(5)5-fluoropyridin-2-amine(15)5-iodopyridin-2-amine(17)5-iodopyridin-2-ol(7)5-methoxy-1H-pyrrolo[2,3-c]pyridine(4)5-methoxypyridin-3-amine(4)5-methylimidazo[1,2-a]pyridine(4)5-methylpyridin-2-amine(7)5-methylpyridin-3-amine(4)5-nitronicotinic acid(5)5-nitropyridin-2-amine(6)5-phenylpicolinic acid(4)6,7-dihydro-5H-cyclopenta[b]pyridine(18)6,7-dihydro-5H-cyclopenta[c]pyridine(7)6,7-dihydro-5H-pyrrolo[3,4-b]pyridine(5)6-(trifluoromethyl)pyridin-2-amine(11)6-(trifluoromethyl)pyridin-3-amine(5)6-bromo-1H-pyrazolo[4,3-b]pyridine(4)6-bromo-1H-pyrrolo[3,2-b]pyridine(7)6-bromo-1H-pyrrolo[3,2-c]pyridine(5)6-bromoimidazo[1,2-a]pyridine(14)6-bromonicotinaldehyde(8)6-bromonicotinic acid(9)6-bromonicotinonitrile(7)6-bromopicolinic acid(5)6-bromopyrazolo[1,5-a]pyridine(8)6-bromopyridin-2-amine(8)6-bromopyridin-3-amine(12)6-bromopyridin-3-ol(13)6-chloro-1H-pyrazolo[4,3-c]pyridine(8)6-chloro-1H-pyrrolo[3,2-c]pyridine(13)6-chloro-3-nitropyridin-2-amine(4)6-chloro-5-(trifluoromethyl)pyridin-2-amine(3)6-chloro-5-iodopyridin-2-amine(3)6-chloroimidazo[1,2-a]pyridine(4)6-chloronicotinaldehyde(12)6-chloronicotinic acid(4)6-chloropicolinic acid(12)6-chloropicolinonitrile(9)6-chloropyridin-2-amine(21)6-chloropyridin-2-ol(8)6-chloropyridin-3-amine(4)6-chloropyridine-3-sulfonyl chloride(5)6-fluoro-1H-pyrrolo[2,3-b]pyridine(3)6-fluoropyridin-2-amine(8)6-fluoropyridin-3-amine(4)6-methoxy-1H-pyrrolo[2,3-b]pyridine(3)6-methoxynicotinaldehyde(4)6-methoxypicolinic acid(7)6-methoxypyridin-2-amine(14)6-methoxypyridin-3-amine(8)6-methylnicotinic acid(6)6-methylnicotinonitrile(7)6-methylpicolinic acid(11)6-methylpicolinonitrile(10)6-methylpyridin-2-amine(46)6-methylpyridin-2-ol(23)6-methylpyridin-3-amine(5)6-methylpyridin-3-ol(7)6-phenoxypyridin-3-amine(5)6-phenylnicotinic acid(7)6-phenylpicolinic acid(3)7-bromo-1H-pyrrolo[2,3-c]pyridine(4)7-chloro-1H-pyrrolo[2,3-c]pyridine(5)7-methoxy-1H-pyrrolo[2,3-c]pyridine(5)7-methylimidazo[1,2-a]pyridine(3)9H-pyrido[3,4-b]indole(5)bis(pyridin-2-ylmethyl)amine(6)dimethyl pyridine-2,6-dicarboxylate(7)dimethyl(pyridin-2-yl)phosphine oxide(8)dimethyl(pyridin-3-yl)phosphine oxide(7)ethyl 2-(pyridin-2-yl)acetate(11)ethyl 2-methylnicotinate(6)ethyl 4,6-dichloronicotinate(4)ethyl 6-chloropicolinate(7)ethyl isonicotinate(12)ethyl nicotinate(25)ethyl picolinate(41)ethyl pyrazolo[1,5-a]pyridine-3-carboxylate(11)furo[3,2-b]pyridine(7)furo[3,2-c]pyridine(7)imidazo[1,2-a]pyridin-2-ylmethanol(3)imidazo[1,2-a]pyridine-5-carboxylic acid(3)imidazo[1,2-a]pyridine-6-carbonitrile(3)isonicotinaldehyde(35)isonicotinamide(12)isonicotinic acid(23)isonicotinohydrazide(7)isonicotinonitrile(16)methyl 1H-pyrazolo[3,4-b]pyridine-5-carboxylate(3)methyl 1H-pyrrolo[2,3-b]pyridine-2-carboxylate(3)methyl 2-(pyridin-2-yl)acetate(9)methyl 2-chloroisonicotinate(11)methyl 2-chloronicotinate(6)methyl 3-bromopicolinate(9)methyl 3-chloropicolinate(9)methyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)nicotinate(4)methyl 5-hydroxypicolinate(5)methyl 6-bromonicotinate(6)methyl 6-chloropicolinate(11)methyl 6-fluoronicotinate(4)methyl 6-methoxynicotinate(4)methyl isonicotinate(16)methyl nicotinate(52)methyl pyrazolo[1,5-a]pyridine-3-carboxylate(16)N,N-dimethylpyridin-2-amine(10)N,N-dimethylpyridin-4-amine(6)N-(pyridin-2-yl)benzamide(9)N-benzylpyridin-2-amine(9)N-methylpicolinamide(10)N-methylpyridin-2-amine(47)N-methylpyridin-3-amine(15)N-phenyl-4-(pyridin-3-yl)pyrimidin-2-amine(5)N-phenylpicolinamide(8)N-phenylpyridin-2-amine(9)nicotinaldehyde(107)nicotinamide(64)nicotinic acid(53)nicotinonitrile(82)oxazolo[4,5-b]pyridine(7)oxazolo[5,4-b]pyridine(5)phenyl(pyridin-2-yl)methanone(10)phenyl(pyridin-3-yl)methanone(8)picolinaldehyde(82)picolinamide(38)picolinic acid(22)picolinimidamide(13)picolinohydrazide(5)pyrazolo[1,5-a]pyridine-2-carboxylic acid(5)pyrazolo[1,5-a]pyridine-3-carboxylic acid(12)pyridin-2-amine(29)pyridin-2-ol(96)pyridin-2-ylboronic acid(9)pyridin-2-ylmethanamine(38)pyridin-2-ylmethanol(65)pyridin-3-amine(21)pyridin-3-ol(13)pyridin-3-ylboronic acid(49)pyridin-3-ylmethanamine(29)pyridin-3-ylmethanol(39)pyridin-4-amine(22)pyridin-4-ol(16)pyridin-4-ylboronic acid(21)pyridin-4-ylmethanamine(12)pyridin-4-ylmethanol(16)pyridine 1-oxide(67)pyridine-2,3-diamine(5)pyridine-2,6-diamine(12)pyridine-2,6-dicarboxylic acid(9)pyridine-2,6-diol(6)pyridine-2-sulfonamide(6)pyridine-2-sulfonyl chloride(9)pyridine-2-thiol(8)pyridine-3-sulfonic acid(6)pyridine-3-sulfonyl chloride(25)pyrido[2,3-b]pyrazine(12)pyrido[2,3-d]pyrimidine(8)pyrido[3,2-d]pyrimidine(14)pyrido[3,4-b]pyrazine(11)pyrido[3,4-d]pyrimidine(10)tert-butyl (2-chloropyridin-4-yl)carbamate(4)tert-butyl 1H-pyrazolo[3,4-b]pyridine-1-carboxylate(7)tert-butyl 4-(pyridin-2-yl)piperazine-1-carboxylate(12)tert-butyl pyridin-2-ylcarbamate(37)tert-butyl pyridin-3-ylcarbamate(16)tert-butyl pyridin-4-ylcarbamate(6)thiazolo[4,5-b]pyridine(12)thiazolo[5,4-b]pyridine(13)thiazolo[5,4-c]pyridine(5)thieno[2,3-b]pyridine(22)thieno[2,3-c]pyridine(14)thieno[3,2-b]pyridine(18)thieno[3,2-c]pyridine(13)2-Pyridone Derivatives(198)acetylpyridin(5)aminopicolinate(8)aminopicolinic(23)aminopyridine(62)benzyloxypyridine(36)bipyridine(202)bromo-picolin-amide(9)bromopicolinaldehyde(18)bromopicolinate(19)bromopicolinic(4)bromopicolinic acid(4)bromopicolinonitrile(6)bromopyridine(209)bromopyrido(13)bromotriazolopyridine(25)butylpyridine(5)carbazo-pyridine(4)chloro-pyrrolopyridine-carboxylate(19)chloro-triazolopyridine(29)chloroisonicotinic(8)chloroisonicotinic acid(1)chloronicotinaldehyde(23)chloronicotinamide(11)chloronicotinate(45)chloronicotinic(41)chloropyridine(254)cyanopicolinate(7)cyanopicolinic(6)cyanopyridine(34)cyclopropoxypyridine(6)cyclopropylpyridine(30)dibromopicolinate(4)dibromopicolinic(6)dibromopicolinic acid(3)dibromopyridine(36)dichloropicolinate(9)dichloropicolinic(3)dichloropicolinonitrile(4)dichloropyridine(120)difluoromethyl-pyridine(4)difluoronicotinic(3)difluoropicolinic(4)difluoropyridine(58)dihydropyridine(128)dihydropyrido(27)dihydroxy-pyridine(14)dihydroxypyridine(3)diiodopyridine(13)dimethoxypicolinic(8)dimethoxypyridine(33)dimethylpyridine(143)diphenylpyridine(6)dipicolinamide(4)dipyridine(28)ethoxypicolinic(8)ethoxypicolinic acid(2)ethoxypyridine(35)ethylpyridine(36)ethynylpicolinate(3)ethynylpyridine(23)fluoro-pyridine--boronate(30)fluoro-pyridine-borolane(67)fluoro-triazolopyridine(4)fluoro-trifluoromethyl-pyridine(606)fluoropicolinaldehyde(8)fluoropicolinamide(8)fluoropicolinate(22)fluoropyridine(288)fluoropyrido(3)formylpicolinate(4)formylpyridine(23)furopyridine(31)hexahydropyrido(7)hydrazinylpyridine(22)hydroxymethylpyridine(27)hydroxypicolinate(13)hydroxypicolinic(9)hydroxypicolinonitrile(7)hydroxypyridine(33)imidazol-pyridine(98)imidazopyridin-amine(40)imidazopyridine(627)iodo-pyrazolopyridine(32)iodopicolinate(5)iodopicolinic(13)iodopicolinic acid(4)iodopyridine(167)isonicotinic(8)isopropoxypyridine(20)isopropylpyridine(32)methoxy-pyridine-boronate(38)methoxy-pyrrolopyridine(45)methoxy-trifluoromethyl-pyridine(37)methoxyisonicotinaldehyde(10)methoxynicotinaldehyde(18)methoxypicolinaldehyde(13)methoxypicolinamide(13)methoxypicolinate(21)methoxypicolinic(20)methoxypicolinic acid(4)methoxypicolinimidamide(5)methoxypicolinonitrile(13)methoxypyridine(342)methyl-imidazopyridine(84)methyl-pyrazolopyridine(143)methyl-pyrrolopyridine(209)methyl-triazolopyridine(5)methylisonicotinamide(8)methylisonicotinate(14)methylnicotinaldehyde(30)methylnicotinamide(13)methylpicolinaldehyde(16)methylpicolinamide(15)methylpicolinate(27)methylpicolinic(23)methylpicolinonitrile(28)methylpyridine(647)methylpyrido(13)nitropyridine(393)oxadiazo-pyridine(4)oxopyridin(2)phenoxypyridin(56)phenylpicolinate(3)phenylpyridine(56)picolinate(42)picoline-boronate(5)picolinic(30)piperazine-pyridine(145)pyranopyridine(4)pyrazol-pyridine(513)pyrazolopyridin-ol(8)pyrazolopyridine(7)pyridin-benzene(554)pyridin-ethanamine(93)pyridin-methanamine(136)pyridin-piperidine(169)pyridin-pyrimidin(124)pyridinamine(4)pyridine-boronate(187)pyridine-hydrochloride(29)pyridine-propylamine(7)pyridine-pyrazole(513)pyridine-pyrrole(36)pyridine-pyrrolidine(112)pyridine-thiazol-amide(23)pyridine-thiazole(135)pyridineboronic acid(4)pyridinecarbonitrile(50)pyridinecarboxylate(13)pyridinecarboxylic(18)pyridinemethanol(8)pyridinoindole(25)pyridinone(6)pyridooxazin-one(1)pyridopyrazine(39)pyridopyrimidin(183)pyridoxazine(37)pyridyl-benzene-boronic acid(26)pyridyl-benzene-boronic acid/ester(3)pyridyl-ethanol(49)pyridyl-phenyl-boronate(2)pyridylmethyl(6)pyrrolopyridine(43)pyrrolopyridine-carboxylate(70)pyrrolopyrimidin-amine(80)terpyridine(32)tetrachloropyridine(5)tetrafluoropyridine(21)tetrahydropyrazolopyridine(8)tetrahydropyridine(49)tetrahydropyrido(32)thiazolopyridine(68)thienopyridine(132)triazol-pyridine(4)triazolopyridin-amine(41)triazolopyridine(51)triazolopyridine-carboxylate(16)triazolopyridine-carboxylic(6)tribromopyridine(9)trichloropyridine(17)trichloropyrido(3)trifluoromethyl-pyridin-amine(133)trifluoromethyl-pyridine-ethylamine(4)trifluoromethylpyridine(6)trifluoropyridine(8)trimethylpyridine(5)
Pyrimidines(14550)
(R)-7-(tetrahydrofuran-2-yl)-7H-pyrrolo[2,3-d]pyrimidine(8)(R)-N-(piperidin-3-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine(5)1,4,5,6-tetrahydropyrimidine(4)1-(3-fluorotetrahydrofuran-2-yl)pyrimidin-2(1H)-one(6)1-(5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione(31)1-(pyrimidin-2-yl)ethan-1-one(5)1-(pyrimidin-4-yl)ethan-1-one(6)1-(tetrahydrofuran-2-yl)pyrimidin-2(1H)-one(47)1H-pyrazolo[3,4-d]pyrimidin-4-amine(5)1H-pyrazolo[4,3-d]pyrimidine(5)2,4,5-trichloropyrimidine(6)2,4,6-trichloropyrimidine(14)2,4,6-triphenylpyrimidine(9)2,4-dichloro-6-methylpyrimidine(7)2,4-dichloropyrimidin-5-amine(6)2,4-dimethoxypyrimidine(13)2,4-dimethylpyrimidine(7)2-(1H-pyrazol-1-yl)pyrimidine(6)2-(chloromethyl)pyrimidine(5)2-(methylsulfonyl)pyrimidine(25)2-(methylthio)pyrimidin-4-amine(10)2-(methylthio)pyrimidine(19)2-(piperazin-1-yl)pyrimidine(11)2-(piperidin-1-yl)pyrimidine(16)2-(propylthio)pyrimidine(5)2-(pyridin-2-yl)pyrimidine(8)2-(pyrrolidin-1-yl)pyrimidine(8)2-(trifluoromethyl)pyrimidine(27)2-aminopyrimidin-4(3H)-one(11)2-bromopyrimidine(22)2-chloro-4-(trifluoromethyl)pyrimidine(6)2-chloro-4-methoxypyrimidine(14)2-chloro-4-methylpyrimidine(16)2-chloro-4-phenylpyrimidine(7)2-chloro-5-fluoropyrimidine(6)2-chloro-5-methylpyrimidine(5)2-chloro-5-nitropyrimidine(5)2-chloro-7H-pyrrolo[2,3-d]pyrimidine(13)2-chloropyrimidin-4-amine(22)2-cyclopropylpyrimidine(10)2-ethoxypyrimidine(7)2-ethylpyrimidine(6)2-fluoropyrimidine(9)2-hydrazineylpyrimidine(6)2-iodopyrimidine(8)2-methoxypyrimidin-4-amine(6)2-methoxypyrimidine(43)2-methylpyrimidin-4-amine(23)2-methylpyrimidin-4-ol(11)2-phenoxypyrimidine(8)2-phenylpyrimidin-5-amine(5)3-methylpyrimidin-4(3H)-one(6)4,5-dichloropyrimidine(7)4,6-dichloro-2-(methylthio)pyrimidine(9)4,6-dichloro-2-methylpyrimidine(7)4,6-dichloropyrimidin-2-amine(9)4,6-dichloropyrimidine(14)4,6-diphenylpyrimidine(7)4-(dimethoxymethyl)pyrimidine(5)4-(piperazin-1-yl)pyrimidine(13)4-(piperidin-1-yl)pyrimidine(9)4-(pyridin-3-yl)pyrimidine(5)4-(pyrimidin-2-yl)morpholine(9)4-(pyrimidin-4-yl)morpholine(13)4-(trifluoromethyl)pyrimidin-2-amine(6)4-(trifluoromethyl)pyrimidine(12)4-amino-1-(tetrahydrofuran-2-yl)pyrimidin-2(1H)-one(22)4-bromopyrimidine(24)4-chloro-2-(methylthio)pyrimidine(29)4-chloro-2-methoxypyrimidine(12)4-chloro-2-phenylpyrimidine(5)4-chloro-5-fluoro-2-methylpyrimidine(5)4-chloro-5-iodopyrimidine(6)4-chloro-5-methoxypyrimidine(7)4-chloro-5-methylpyrimidine(8)4-chloro-6-methoxypyrimidine(5)4-chloropyrimidin-2-amine(23)4-chloropyrimidin-5-amine(7)4-chloropyrimidine-5-carbonitrile(6)4-ethoxypyrimidine(6)4-fluoropyrimidine(8)4-iodopyrimidine(9)4-methoxy-2-methylpyrimidine(8)4-methoxypyrimidin-2-amine(6)4-methoxypyrimidine(50)4-methyl-2-(methylthio)pyrimidine(7)4-methylpyrimidin-2-amine(9)4-methylpyrimidine(22)4-phenoxypyrimidine(5)4-phenylpyrimidin-2-amine(15)5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine(7)5,6,7,8-tetrahydropyrido[4,3-d]pyrimidine(17)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine(27)5-(benzyloxy)pyrimidine(5)5-benzylpyrimidine(7)5-bromo-1-(tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione(4)5-bromo-2-(methylthio)pyrimidine(6)5-bromo-2-chloropyrimidine(6)5-bromo-2-methylpyrimidine(5)5-bromo-4-chloropyrimidine(4)5-bromo-7H-pyrrolo[2,3-d]pyrimidine(3)5-bromopyrimidin-4(3H)-one(3)5-chloropyrimidin-4(3H)-one(4)5-chloropyrimidine(8)5-chloropyrimidine-2,4(1H,3H)-dione(3)5-ethylpyrimidine(5)5-fluoro-1-(tetrahydrofuran-2-yl)pyrimidin-2(1H)-one(7)5-fluoro-1-(tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione(3)5-fluoropyrimidin-4(3H)-one(3)5-fluoropyrimidine(11)5-fluoropyrimidine-2,4(1H,3H)-dione(3)5-iodo-7H-pyrrolo[2,3-d]pyrimidine(4)5-iodopyrimidine(5)5-methylpyrimidine(3)5-phenylpyrimidine(16)5H-pyrrolo[3,2-d]pyrimidine(16)6,7-dihydro-5H-cyclopenta[d]pyrimidine(5)6,7-dihydro-5H-pyrrolo[3,4-d]pyrimidine(8)6-chloro-1H-pyrazolo[3,4-d]pyrimidine(12)6-chloropyrimidin-4-amine(8)7-(phenylsulfonyl)-7H-pyrrolo[2,3-d]pyrimidine(6)7H-pyrrolo[2,3-d]pyrimidin-2-amine(5)7H-pyrrolo[2,3-d]pyrimidin-4-amine(3)[1,2,4]triazolo[1,5-a]pyrimidine(14)ethyl 4-chloropyrimidine-5-carboxylate(5)ethyl pyrazolo[1,5-a]pyrimidine-3-carboxylate(6)ethyl pyrimidine-4-carboxylate(7)ethyl pyrimidine-5-carboxylate(5)imidazo[1,2-c]pyrimidine(6)methyl pyrimidine-2-carboxylate(10)methyl pyrimidine-4-carboxylate(11)methyl pyrimidine-5-carboxylate(5)N,N-dimethylpyrimidin-2-amine(10)N-(9-((2R,5S)-5-((trityloxy)methyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)benzamide(5)N-benzyl-9H-purin-6-amine(6)N-cyclopropylpyrimidin-2-amine(5)N-methyl-N-(4-phenylpyrimidin-2-yl)methanesulfonamide(7)N-methylpyrimidin-2-amine(10)N-methylpyrimidin-4-amine(10)N-phenyl-4-(pyridin-3-yl)pyrimidin-2-amine(5)N-phenylpyrimidin-2-amine(5)N-phenylpyrimidin-4-amine(7)N2,N4-diphenylpyrimidine-2,4-diamine(5)pyrido[2,3-d]pyrimidine(8)pyrido[3,2-d]pyrimidine(14)pyrido[3,4-d]pyrimidine(10)pyrimidin-2-ol(21)pyrimidin-2-ylmethanamine(7)pyrimidin-2-ylmethanol(5)pyrimidin-4-amine(29)pyrimidin-4-ol(25)pyrimidin-5-amine(6)pyrimidin-5-ol(5)pyrimidin-5-ylboronic acid(9)pyrimidine-2,4-diamine(14)pyrimidine-2-carbonitrile(14)pyrimidine-2-carboxylic acid(11)pyrimidine-2-thiol(6)pyrimidine-4,6(1H,5H)-dione(3)pyrimidine-4-carbonitrile(9)pyrimidine-4-carboxylic acid(17)pyrimidine-5-carbaldehyde(21)pyrimidine-5-carbonitrile(6)pyrimidine-5-carboxylic acid(7)tetrahydropyrimidin-2(1H)-one(6)thiazolo[5,4-d]pyrimidine(5)thieno[2,3-d]pyrimidine(30)thieno[3,2-d]pyrimidine(24)allylpyrimidine(3)amino-pyrazolopyrimidine(6)amino-pyrrolopyrimidine(50)amino-triazolopyrimidine(4)aminopyrimidine(37)benzofuranopyrimidine(4)bipyrimidine(4)bromo-pyrazolopyrimidine(44)bromo-pyrrolopyrimidine(28)bromo-trifluoromethyl-pyrimidine(15)bromopyrimidine(47)butylpyrimidine(59)chloro-pyrazolopyrimidine(76)chloro-pyrrolopyrimidine(102)chloro-triazolopyrimidine(6)chloro-trifluoromethyl-pyrimidine(40)chloropyrimidine(86)cyclopropylpyrimidine(18)diaminopyrimidine(23)dibromopyrimidine(15)dichloropyrimidine(38)difluoropyrimidine(6)dihydropyrimidine(35)dihydroxypyrimidine(5)dimethoxypyrimidine(22)dimethylpyrimidine(63)dioxaborolan-pyrimidine(47)diphenylpyrimidine(7)ethoxypyrimidine(18)ethylpyrimidine(16)fluoropyrimidine(32)furanopyrimidine(12)hydrazinylpyrimidine(3)hydroxy-pyrazolopyrimidine(6)hydroxypyrimidine(19)imidazolyl-pyrimidine(2)imidazopyrimidine(84)iodo-pyrrolopyrimidine(17)iodopyrimidine(28)isopropoxypyrimidine(1)isopropylpyrimidine(14)mercaptopyrimidine(2)methoxypyrimidine(107)methyl-triazolopyrimidine(16)methylpyrimidine(212)methyluracil(2)nitropyrimidine(24)phenoxypyrimidine(43)phenylpyrimidine(29)piperazin-pyrimidin(6)piperidine-pyrimidine(192)propylpyrimidine(1)pyrazolo-pyrimidine(261)pyrazolopyrimid-one(28)pyrazolopyrimidine(261)pyridin-pyrimidin(124)pyridopyrimidin(183)pyrimidinamine(2)pyrimidine-boronic acid(37)pyrimidine-boronic-acid(1)pyrimidine-hydrochloride(105)pyrimidine-methanol(46)pyrimidine-morpholine(65)pyrimidinecarbonitrile(11)pyrimidinecarboxylate(4)pyrimidinecarboxylic(3)pyrimidinone(9)pyrimido-indole(5)pyrrolidine-pyrimidine(41)pyrrolopyrimidin-one(22)pyrrolopyrimidine(226)pyrrolopyrimidine-carboxylate(19)tetrahydrofuran-pyrimidine(103)tetrahydropyrimidine(11)tetraoxatetradecan(10)thiazopyrimidine(50)thienopyrimidine(127)triazolopyrimidine(41)trichloropyrimidine(11)trifluoromethyl-phenyl-pyrimidin(35)trifluoromethyl-pyrimidin-amine(59)trifluoromethyl-pyrimidin-ol(4)trifluoromethylpyrimidine(2)
Pyrroles(4966)
(9H-fluoren-9-yl)methyl pyrrolidine-1-carboxylate(26)(Z)-3-((1H-pyrrol-2-yl)methylene)indolin-2-one(8)1,3-dihydro-2H-pyrrolo[2,3-b]pyridin-2-one(19)1,3-dihydro-2H-pyrrolo[3,2-b]pyridin-2-one(11)1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole(6)1-(1H-pyrrolo[2,3-b]pyridin-3-yl)ethan-1-one(4)1-(phenylsulfonyl)-1H-pyrrole(12)1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridine(30)1-benzylpyrrolidin-2-one(18)1-methyl-1H-pyrrole(29)1H-pyrrol-1-amine(6)1H-pyrrole-2-carbaldehyde(17)1H-pyrrole-2-carbonitrile(6)1H-pyrrole-2-carboxylic acid(28)1H-pyrrole-3-carbaldehyde(7)1H-pyrrole-3-carbonitrile(8)1H-pyrrolo[2,3-b]pyridin-6-amine(4)1H-pyrrolo[2,3-b]pyridine-3-carboxylic acid(5)1H-pyrrolo[2,3-c]pyridin-7-ol(3)2,3-dihydro-1H-pyrrolo[2,3-b]pyridine(10)2,3-dihydro-1H-pyrrolo[3,4-c]pyridine(6)2-bromo-1H-pyrrole(10)2-methyl-1H-pyrrole(32)2-nitro-1H-pyrrole(3)2-phenyl-1H-pyrrole(24)3,6-di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione(17)3-bromo-1H-pyrrole(8)3-bromo-1H-pyrrolo[2,3-b]pyridine(7)3-iodo-1H-pyrrolo[3,2-c]pyridine(4)3-methyl-1H-pyrrolo[2,3-b]pyridine(5)3-phenyl-1H-pyrrole(5)3-phenylpyrrolidine(26)4-bromo-1H-pyrrolo[2,3-b]pyridine(9)4-chloro-1H-pyrrolo[2,3-b]pyridine(13)4H-thieno[3,2-b]pyrrole(6)5-bromo-1H-pyrrolo[2,3-b]pyridine(13)5-chloro-1H-pyrrolo[2,3-c]pyridine(5)5-methoxy-1H-pyrrolo[2,3-c]pyridine(4)5H-pyrrolo[2,3-b]pyrazine(14)5H-pyrrolo[3,2-d]pyrimidine(16)7-(phenylsulfonyl)-7H-pyrrolo[2,3-d]pyrimidine(6)7-methoxy-1H-pyrrolo[2,3-c]pyridine(5)ethyl 1H-pyrrole-2-carboxylate(24)ethyl 1H-pyrrole-3-carboxylate(8)methyl 1H-pyrrole-2-carboxylate(22)methyl 1H-pyrrole-3-carboxylate(5)methyl 1H-pyrrolo[2,3-b]pyridine-3-carboxylate(4)methyl 1H-pyrrolo[2,3-b]pyridine-6-carboxylate(3)pyrrolo[2,1-f][1,2,4]triazine(14)tert-butyl 1H-pyrrole-1-carboxylate(8)tert-butyl 1H-pyrrole-2-carboxylate(3)amino-pyrrole-carboxylate(162)amino-pyrrolopyrimidine(50)bromo-pyrrolopyridine(169)bromo-pyrrolopyridine-carboxylate(20)bromopyrrole-carboxylate(61)bromopyrrolo(8)chloro-pyrrolopyridine(33)chloro-pyrrolopyridine-carboxylate(19)chloropyrrolo(10)dihydropyrrolo(15)dimethyl-pyrrole(249)fluoro-pyrrolopyridine(64)fluoropyrrolo(2)formyl-pyrrole-carboxylate(14)furopyrrole(6)iodo-pyrrolopyridine(39)iodo-pyrrolopyridine-carboxylate(3)methoxy-pyrrolopyridine(45)methyl-pyrrolopyridine(209)methylpyrrole(3)nitro-pyrrolopyridine(15)octahydropyrrolo(6)phenyl-pyrrole(75)pyridine-pyrrole(36)pyrrol-amine(165)pyrrole-carbonitrile(67)pyrrole-carboxylate(43)pyrrole-carboxylic acid(40)pyrrolizine-carboxylate(16)pyrroloimidazole(13)pyrrolopyrazine(47)pyrrolopyrazole(51)pyrrolopyridazine(3)pyrrolopyridin-one(63)pyrrolopyridine(43)pyrrolopyridine-carbonitrile(24)pyrrolopyridine-carboxylate(70)pyrrolopyridine-carboxylic acid(45)pyrrolopyridine-hydroxy(2)pyrrolopyridine-methanamine(21)pyrrolopyridine-methanol(3)pyrrolopyridine-methylamine(3)pyrrolopyrimidin-amine(80)pyrrolopyrimidin-one(22)pyrrolopyrimidine(226)pyrrolopyrimidine-carboxylate(19)pyrrolopyrimidine-carboxylic acid(15)pyrrolopyrrole(40)thieno-pyrrole(41)trifluoromethyl-pyrrolo(26)
Pyrrolidines(14855)
(3aR,6aS)-octahydrocyclopenta[c]pyrrole(5)(9H-fluoren-9-yl)methyl pyrrolidine-1-carboxylate(26)(R)-3-(pyrrolidin-2-yl)pyridine(9)(R)-3-benzylpyrrolidine(6)(R)-3-phenylpyrrolidine(8)(S)-3-phenylpyrrolidine(14)(S)-N-(((S)-tetrahydro-2H-pyran-2-yl)methyl)pyrrolidine-2-carboxamide(4)(S)-N-(4-(thiazol-5-yl)benzyl)pyrrolidine-2-carboxamide(7)(S)-N-phenethyl-3-(pyrrolidin-2-yl)propanamide(5)1-((benzyloxy)carbonyl)pyrrolidine-3-carboxylic acid(6)1-(3-chlorophenyl)pyrrolidine(3)1-(phenylsulfonyl)pyrrolidine(15)1-(pyrrolidin-1-yl)ethan-1-one(9)1-benzyl 2-methyl pyrrolidine-1,2-dicarboxylate(7)1-benzylpyrrolidin-2-one(18)1-benzylpyrrolidine(26)1-methylpyrrolidin-2-one(13)1-phenylpyrrolidin-2-one(18)2,2-dimethylpyrrolidine(3)2,5-dioxopyrrolidin-1-yl ((benzyloxy)carbonyl)glycinate(8)2,5-dioxopyrrolidin-1-yl benzoate(8)2,8-diazaspiro[4.5]decan-3-one(6)2-((3-phenylpropyl)amino)-1-(pyrrolidin-1-yl)ethan-1-one(2)2-(2-chlorophenyl)pyrrolidine(17)2-(2-fluorophenyl)pyrrolidine(16)2-(3-fluorophenyl)pyrrolidine(5)2-(methoxymethyl)pyrrolidine(6)2-(pyrrolidin-1-yl)benzoic acid(3)2-(pyrrolidin-1-yl)pyridine(11)2-(pyrrolidin-1-yl)pyrimidine(8)2-(pyrrolidin-2-yl)acetic acid(8)2-(pyrrolidin-2-yl)pyridine(5)2-(pyrrolidin-3-yl)acetic acid(6)2-amino-1-(pyrrolidin-1-yl)ethan-1-one(5)2-azabicyclo[2.2.1]heptane(6)2-methyl-1-phenyl-1H-pyrrole(8)2-methylpyrrolidine(20)3,3-difluoropyrrolidine(27)3-(pyrrolidin-2-yl)pyridine(6)3-aminopyrrolidin-2-one(5)3-bromopyrrolidine(6)3-hydroxypyrrolidin-2-one(4)3-phenoxypyrrolidine(5)3-phenyl-1-(pyrrolidin-1-yl)propan-1-one(5)3-phenylpyrrolidine(26)4-(hydroxymethyl)pyrrolidin-2-one(3)4-(pyrrolidin-1-yl)aniline(3)4-(pyrrolidin-1-yl)piperidine(6)4-hydroxypyrrolidin-2-one(7)4-phenylpyrrolidin-2-one(7)4H-thieno[3,4-c]pyrrole-4,6(5H)-dione(7)5-(aminomethyl)pyrrolidin-2-one(3)5-(hydroxymethyl)pyrrolidin-2-one(7)5-azaspiro[2.4]heptane(13)5-oxopyrrolidine-2-carboxylic acid(4)6-azaspiro[3.4]octane(4)benzyl (S)-pyrrolidin-3-ylcarbamate(7)benzyl 3-hydroxypyrrolidine-1-carboxylate(5)benzyl-2-(hydroxymethyl)pyrrolidine-1-carboxylate(9)benzyl-3-aminopyrrolidine-1-carboxylate(7)ethyl 5-oxopyrrolidine-2-carboxylate(3)hexahydro-1H-pyrrolizine(12)methyl 5-oxopyrrolidine-2-carboxylate(3)methyl prolinate(43)methyl pyrrolidine-3-carboxylate(9)N,N-dimethylpyrrolidin-3-amine(7)N-methylpyrrolidin-3-amine(6)octahydrocyclopenta[c]pyrrole(6)phenyl(pyrrolidin-1-yl)methanone(12)proline(13)pyrrolidin-2-ylmethanamine(12)pyrrolidin-2-ylmethanol(26)pyrrolidin-3-ol(50)pyrrolidin-3-ylmethanamine(6)pyrrolidin-3-ylmethanol(13)pyrrolidine-2-carbonitrile(9)pyrrolidine-2-carboxamide(12)pyrrolidine-3-carbonitrile(6)pyrrolidine-3-carboxylic acid(14)tert-butyl (2-oxopyrrolidin-3-yl)carbamate(3)tert-butyl (5-oxopyrrolidin-3-yl)carbamate(3)tert-butyl 2-oxopyrrolidine-1-carboxylate(15)tert-butyl 3-((methylsulfonyl)oxy)pyrrolidine-1-carboxylate(5)tert-butyl 3-hydroxypyrrolidine-1-carboxylate(20)tert-butyl 3-methoxypyrrolidine-1-carboxylate(6)tert-butyl pyrrolidin-3-ylcarbamate(12)tert-butyl pyrrolidine-1-carboxylate(21)acetylpyrrolidine(9)aminopyrrolidine(28)benzylpyrrolidine(33)bipyrrolidine(45)bromopyrrolidine(7)carbamoylpyrrolidine(5)cyanopyrrolidine(8)difluoropyrrolidine(16)dihydroxypyrrolidine(3)dimethylpyrrolidine(31)dioxaborolane-pyrrolidine(3)dioxopyrrolidin(131)ethylpyrrolidine(9)fluoropyrrolidine(26)formylpyrrolidine(10)hydroxypyrrolidine(96)mercaptopyrrolidine(4)methoxypyrrolidine(19)methylenepyrrolidine(2)methylpyrrolidine(129)phenoxypyrrolidine(3)phenylpyrrolidine(22)pyridine-pyrrolidine(112)pyrrolidine-benzene-boronic acid(27)pyrrolidine-benzene-boronic acid/ester(4)pyrrolidine-carboxylate(31)pyrrolidine-carboxylic acid(269)pyrrolidine-hydrochloride(316)pyrrolidine-methanol(18)pyrrolidine-pyrimidine(41)pyrrolidinone(59)trifluoromethyl-phenyl-pyrrolidine(5)
Pyrrolines(1758)
1,3-dihydro-2H-pyrrolo[2,3-c]pyridin-2-one(5)1-phenyl-1H-pyrrole-2,5-dione(12)2,3-dihydro-1H-pyrrolo[2,3-c]pyridine(5)2,3-dihydro-1H-pyrrolo[3,2-c]pyridine(7)2,5-dihydro-1H-pyrrol-2-one(5)2,5-dihydro-1H-pyrrole(13)2-phenyl-2,5,6,7-tetrahydropyrrolo[2,1-c][1,2,4]triazol-4-ium(6)3,4-dihydro-2H-pyrrole(5)3-bromo-1H-pyrrole-2,5-dione(3)5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole(8)6,7-dihydro-5H-pyrrolo[1,2-a]imidazole(9)6,7-dihydro-5H-pyrrolo[3,4-b]pyridine(5)6,7-dihydro-5H-pyrrolo[3,4-d]pyrimidine(8)
Quinazolines(3145)
2,4-dichloroquinazoline(19)2-(trifluoromethyl)quinazoline(4)2-bromoquinazoline(4)2-chloroquinazolin-4(3H)-one(4)2-cyclopropylquinazoline(5)2-mercaptoquinazolin-4(3H)-one(3)2-methylquinazoline(20)4-chloroquinazoline(39)4-methylquinazoline(4)5,6,7,8-tetrahydroquinazoline(8)5-chloroquinazoline(4)6-bromoquinazoline(7)7-bromoquinazolin-4(3H)-one(4)7-fluoroquinazolin-4(3H)-one(4)7-methoxy-N-phenylquinazolin-4-amine(12)8-bromoquinazoline(8)8-chloroquinazoline(5)8-fluoroquinazoline(4)8-methoxyquinazoline(3)8-methylquinazoline(3)ethyl quinazoline-2-carboxylate(4)N-(3-chlorophenyl)quinazolin-4-amine(10)quinazolin-2-amine(13)quinazolin-2-ol(7)quinazolin-4-amine(8)quinazolin-4-ol(10)amino-quinazoline(93)bromoquinazoline(19)chloroquinazoline(30)dichloroquinazoline(14)dihydroquinazoline(31)dimethoxyquinazoline(18)dimethylquinazoline(5)fluoroquinazoline(20)iodoquinazoline(10)methoxyquinazoline(42)methylquinazoline(46)nitroquinazoline(20)phenylquinazolin(5)quinazolin-one(6)Quinazoline Derivatives(125)quinazoline-carboxylate(4)quinazoline-carboxylic-acid(4)tetrahydroquinazoline(5)trichloroquinazoline(5)trifluoromethyl-quinazoline(17)
Quinolines(11901)
1,2-dihydroquinoline(5)1-cyclopropyl-3-(4-(quinolin-4-yloxy)phenyl)urea(6)2,2'-biquinoline(5)2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinoline(8)2-(quinolin-2-yl)-4,5-dihydrooxazole(7)2-hydroxyquinolin-4(1H)-one(3)2-phenylquinoline(19)3,4-dihydroisoquinoline(11)4-oxo-1,4-dihydroquinoline-3-carboxylic acid(3)4-phenoxyquinoline(5)4-phenylquinoline(9)6-bromo-3,4-dihydroquinolin-2(1H)-one(4)7,8-dihydroquinolin-5(6H)-one(5)7-(benzyloxy)quinoline(6)7-methoxy-3,4-dihydroquinolin-2(1H)-one(3)N-phenyl-N-(4-(quinolin-4-yloxy)phenyl)cyclopropane-1,1-dicarboxamide(5)N-phenylquinolin-4-amine(11)acetylquinoline(5)aminoquinoline(47)benzoquinoline(13)biquinoline(3)bromo-trifluoromethyl-quinoline(35)bromoquinoline(119)chloro-trifluoromethyl-quinoline(16)chloroquinoline(138)dibromoquinoline(6)dichloroquinoline(16)difluoroquinoline(9)dihydroquinoline(128)dihydroxyquinoline(13)dimethoxyquinoline(8)dimethylquinoline(20)dioxaborolane-quinoline(29)ethoxyquinoline(10)ethylquinoline(6)fluoroquinoline(112)hydroxyquinoline(68)imidazoquinoline(2)iodoquinoline(32)methoxyquinoline(157)methylquinoline(182)nitroquinoline(76)phenylquinoline(15)piperazine-quinoline(49)pyranoquinoline(5)quinolin-boronic acid(42)quinoline-carbonitrile(94)quinoline-carboxylate(280)quinoline-carboxylic acid(381)quinoline-ethylamine(5)quinoline-hydrochloride(77)quinolinone(3)trifluoromethyl-hydroxy-quinoline(14)trifluoromethyl-quinolin-amine(33)trifluoromethylquinoline(173)
Quinoxalines(1411)
1,2,3,4-tetrahydroquinoxaline(6)2-chloroquinoxaline(39)2-methylquinoxaline(5)2-phenylquinoxaline(5)3,4-dihydroquinoxalin-2(1H)-one(6)5-bromoquinoxaline(6)6-bromoquinoxaline(9)6-chloroquinoxaline(3)6-methoxyquinoxaline(6)quinoxalin-2-amine(3)quinoxalin-2-ol(4)aminoquinoxalin(1)bromoquinoxaline(8)chloroquinoxaline(14)dichloroquinoxaline(8)dihydroquinoxalin(7)dimethoxyquinoxaline(8)methoxyquinoxaline(9)methylquinoxaline(17)nitroquinoxaline(8)quinoxaline-carboxylate(5)quinoxaline-carboxylic-acid(6)tetrahydroquinoxaline(7)trifluoromethyl-quinoxaline(2)
Spiroes(4714)
1,3-dihydrospiro[indene-2,4'-piperidine](5)1,4-dioxaspiro[4.5]decane(17)2,8-diazaspiro[4.5]decane(5)2-oxa-8-azaspiro[4.5]decane(5)3-phenyloxetane(11)6-azaspiro[3.4]octane(4)7-azaspiro[3.5]nonane(7)9,9'-spirobi[fluorene](19)spiro[cyclopropane-1,3'-indolin]-2'-one(6)spiro[indene-2,4'-piperidin]-1(3H)-one(5)spiro[indoline-3,4'-piperidine](5)aminospiro(4)azaspiro(246)bromospiro(11)diazaspiro(182)dibromospiro(3)dihydrospiro(18)dioxaspiro(39)fluorospiro(4)oxospiro(22)triazaspiro(13)
Tetrahydrofurans(2264)
(2R,3S)-2-((benzoyloxy)methyl)tetrahydrofuran-3-yl benzoate(8)(3aR,6aR)-hexahydrofuro[3,2-b]furan(5)(3aS,6aS)-tetrahydrofuro[3,4-d][1,3]dioxole(6)(R)-7-(tetrahydrofuran-2-yl)-7H-pyrrolo[2,3-d]pyrimidine(8)(R)-N-(9-(tetrahydrofuran-2-yl)-9H-purin-6-yl)benzamide(5)(tetrahydrofuran-2-yl)methanamine(4)(tetrahydrofuran-2-yl)methanol(6)(tetrahydrofuran-3-yl)methanamine(3)1,3-dihydrobenzo[c][1,2]oxaborole(20)1-(3-fluorotetrahydrofuran-2-yl)pyrimidin-2(1H)-one(6)1-(5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione(31)1-(tetrahydrofuran-2-yl)pyrimidin-2(1H)-one(47)1-oxa-8-azaspiro[4.5]decane(5)2-(acetoxymethyl)-5-(9H-purin-9-yl)tetrahydrofuran-3,4-diyl diacetate(6)2-(hydroxymethyl)-5-(9H-purin-9-yl)tetrahydrofuran-3,4-diol(28)2-oxa-8-azaspiro[4.5]decane(5)2-oxabicyclo[2.1.1]hexane(6)3-aminodihydrofuran-2(3H)-one(4)3-iodotetrahydrofuran(3)4-amino-1-(tetrahydrofuran-2-yl)pyrimidin-2(1H)-one(22)4-fluoro-2-(hydroxymethyl)-5-(9H-purin-9-yl)tetrahydrofuran-3-ol(4)4-hydroxydihydrofuran-2(3H)-one(14)5-(hydroxymethyl)dihydrofuran-2(3H)-one(7)5-bromo-1-(tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione(4)5-fluoro-1-(tetrahydrofuran-2-yl)pyrimidin-2(1H)-one(7)5-fluoro-1-(tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione(3)5-oxotetrahydrofuran-2-carboxylic acid(4)9-(tetrahydrofuran-2-yl)-9H-purin-6-amine(30)9-(tetrahydrofuran-2-yl)-9H-purin-6-ol(6)9-(tetrahydrofuran-2-yl)-9H-purine(2)dihydrofuran-3(2H)-one(8)tert-butyl (tetrahydrofuran-3-yl)carbamate(4)tetrahydrofuran-2-carboxylic acid(4)tetrahydrofuran-3-amine(6)tetrahydrofuran-3-carboxylic acid(4)tetrahydrofuran-3-ol(7)aminotetrahydrofuran(7)dihydroxytetrahydrofuran(35)hydroxytetrahydrofuran(16)iodotetrahydrofuran(3)methoxytetrahydrofuran(11)methyltetrahydrofuran(9)oxotetrahydrofuran(26)tetrahydrofuran-carboxylate(26)tetrahydrofuran-carboxylic acid(17)tetrahydrofuran-carboxylic-acid(2)tetrahydrofuran-hydrochloride(16)tetrahydrofuran-methanol(5)tetrahydrofuran-methylamine(15)tetrahydrofuran-pyrimidine(103)
Tetrahydroisoquinolines(1292)
(9H-fluoren-9-yl)methyl 3,4-dihydroisoquinoline-2(1H)-carboxylate(7)1,2,3,4-tetrahydroisoquinolin-5-amine(4)1,2,3,4-tetrahydroisoquinolin-5-ol(3)1,2,3,4-tetrahydroisoquinolin-6-amine(3)1,2,3,4-tetrahydroisoquinolin-6-ol(6)1,2,3,4-tetrahydroisoquinolin-7-amine(3)1,2,3,4-tetrahydroisoquinoline-1-carboxylic acid(19)1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid(12)1,4-dihydroisoquinolin-3(2H)-one(5)1-methyl-1,2,3,4-tetrahydroisoquinoline(4)2-methyl-1,2,3,4-tetrahydroisoquinoline(13)4,4-dimethyl-1,2,3,4-tetrahydroisoquinoline(3)5,6,7,8-tetrahydroisoquinoline(5)5-bromo-1,2,3,4-tetrahydroisoquinoline(4)5-chloro-1,2,3,4-tetrahydroisoquinoline(3)5-fluoro-1,2,3,4-tetrahydroisoquinoline(3)5-methyl-1,2,3,4-tetrahydroisoquinoline(4)6-bromo-1,2,3,4-tetrahydroisoquinoline(4)6-chloro-1,2,3,4-tetrahydroisoquinoline(3)6-fluoro-1,2,3,4-tetrahydroisoquinoline(3)6-methoxy-1,2,3,4-tetrahydroisoquinoline(3)6-nitro-1,2,3,4-tetrahydroisoquinoline(3)7-bromo-1,2,3,4-tetrahydroisoquinoline(4)7-chloro-1,2,3,4-tetrahydroisoquinoline(5)7-fluoro-1,2,3,4-tetrahydroisoquinoline(3)7-methoxy-1,2,3,4-tetrahydroisoquinoline(6)7-nitro-1,2,3,4-tetrahydroisoquinoline(3)isoquinoline-1,3(2H,4H)-dione(7)hydroxy-tetrahydroisoquinolin(3)methyl-tetrahydroisoquinoline(4)other-tetrahydroisoquinoline(51)phenyl-tetrahydroisoquinoline(15)tetrahydroisoquinolin-amine(5)tetrahydroisoquinoline-bromide(6)tetrahydroisoquinoline-carboxylate(16)tetrahydroisoquinoline-carboxylic acid(48)tetrahydroisoquinoline-chloride(44)tetrahydroisoquinoline-hydrochloride(61)tetrahydroisoquinoline-methanol(2)tetrahydroisoquinoline-nitro(6)trifluoromethyl-tetrahydroisoquinoline(4)
Tetrahydropyrans(3342)
(1S,4aR,7aR)-1-(((S)-tetrahydro-2H-pyran-2-yl)oxy)-1,4a,5,7a-tetrahydrocyclopenta[c]pyran(4)(6-phenoxytetrahydro-2H-pyran-2-yl)methanol(19)(R)-2-phenoxytetrahydro-2H-pyran(9)(S)-2-(phenylthio)tetrahydro-2H-pyran(5)(S)-7-(((S)-tetrahydro-2H-pyran-2-yl)oxy)-7,8,9,10-tetrahydrotetracene-5,12-dione(5)(S)-N-(((S)-tetrahydro-2H-pyran-2-yl)methyl)pyrrolidine-2-carboxamide(4)1-(tetrahydro-2H-pyran-4-yl)-1H-pyrazole(6)2-(phenoxymethyl)tetrahydro-2H-pyran(5)2-phenoxytetrahydro-2H-pyran(6)2-phenoxytetrahydro-2H-pyran-4-yl acetate(6)4-phenoxytetrahydro-2H-pyran(5)9-bromoanthracene(5)aminotetrahydropyran(9)chroman-amine(70)difluorochroman(16)fluorochroman(27)hydroxymethyl-tetrahydropyran(3)tetrahydro-carboxylate(144)tetrahydro-pyran-amine(160)tetrahydro-pyran-phenyl(65)tetrahydropyran-carboxylic-acid(4)tetrahydropyran-hydrochloride(36)tetrahydropyran-hydroxy(346)tetrahydropyran-indazole(4)tetrahydropyran-one(109)tetrahydropyran-pyrazole(6)tetrahydropyranopyrazole(4)
Tetrahydroquinolines(1464)
1-ethyl-1,2,3,4-tetrahydroquinoline(6)1-methyl-1,2,3,4-tetrahydroquinoline(6)2,3-dihydroquinolin-4(1H)-one(19)2-methyl-1,2,3,4-tetrahydroquinoline(3)4,4-dimethyl-1,2,3,4-tetrahydroquinoline(3)4-methyl-1,2,3,4-tetrahydroquinoline(2)5,6,7,8-tetrahydroquinoline(17)6-bromo-1,2,3,4-tetrahydroquinoline(6)6-chloro-1,2,3,4-tetrahydroquinoline(3)6-fluoro-1,2,3,4-tetrahydroquinoline(3)7-fluoro-1,2,3,4-tetrahydroquinoline(3)tert-butyl 3,4-dihydroquinoline-1(2H)-carboxylate(11)hydroxy-tetrahydroquinoline(7)tetrahydroquinolin-amine(11)tetrahydroquinoline-benzene(2)tetrahydroquinoline-bromide(14)tetrahydroquinoline-carbonitrile(3)tetrahydroquinoline-carboxylate(3)tetrahydroquinoline-carboxylic acid(18)tetrahydroquinoline-chloride(7)tetrahydroquinoline-fluoro(11)tetrahydroquinoline-hydrochloride(22)tetrahydroquinoline-methoxy(5)tetrahydroquinoline-methyl(42)tetrahydroquinoline-nitro(9)
Thiadiazoles(2120)
1,2,3-thiadiazole(8)1,2,4-thiadiazole(16)1,2,5-thiadiazole(6)1,3,4-thiadiazol-2-amine(9)2-bromo-1,3,4-thiadiazole(11)2-methyl-1,3,4-thiadiazole(12)2-phenyl-1,3,4-thiadiazole(19)4,7-diphenylbenzo[c][1,2,5]thiadiazole(6)4-bromobenzo[c][1,2,5]thiadiazole(14)benzo[d][1,2,3]thiadiazole(7)amino-thiadiazole(24)benzothiadiazole(6)bromo-thiadiazole(58)chloro-thiadiazole(29)methyl-thiadiazole(58)thiadiazol-amine(37)thiadiazole-carboxylate(14)thiadiazole-carboxylic acid(5)thiadiazole-methanol(3)
Thiazoles(6645)
1-(thiazol-2-yl)ethan-1-one(9)2,4-dibromothiazole(5)2,4-dichlorothiazole(4)2,4-dimethylthiazole(7)2,5-dibromothiazole(5)2,5-dimethylthiazole(4)2-(chloromethyl)thiazole(5)2-(methylthio)thiazole(5)2-(thiazol-4-yl)acetic acid(7)2-(trifluoromethyl)thiazole(11)2-bromo-4-methylthiazole(7)2-bromo-5-methylthiazole(7)2-chloro-4-methylthiazole(5)2-chloro-5-methylthiazole(5)2-chlorothiazole(32)2-imino-N-((6R,7R)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-7-yl)-2-(thiazol-4-yl)acetamide(3)2-isopropylthiazole(11)2-methyl-4-phenylthiazole(7)2-phenoxythiazole(5)2-phenylbenzo[d]thiazole(16)2-phenylthiazole-4-carbaldehyde(9)4,5,6,7-tetrahydrobenzo[d]thiazole(14)4,5,6,7-tetrahydrothiazolo[5,4-c]pyridine(10)4,5-dihydrothiazole(8)4-(4-fluorophenyl)thiazole(9)4-(trifluoromethyl)thiazole(7)4-bromothiazole(10)4-cyclopropylthiazole(6)4-methyl-2-phenylthiazole(6)4-methylthiazol-2-amine(7)4-methylthiazole(5)4-phenylthiazol-2-amine(33)5,6-dihydrobenzo[d]thiazol-7(4H)-one(5)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiazole(7)5-bromothiazol-2-amine(6)5-bromothiazole(8)5-methyl-4-phenylthiazole(5)5-methylthiazol-2-amine(6)5-phenylthiazole(20)ethyl 2-aminothiazole-4-carboxylate(6)ethyl 2-aminothiazole-5-carboxylate(5)ethyl 2-phenylthiazole-4-carboxylate(12)ethyl 2-phenylthiazole-5-carboxylate(9)ethyl thiazole-2-carboxylate(7)ethyl thiazole-4-carboxylate(7)ethyl thiazole-5-carboxylate(7)methyl 2-aminothiazole-4-carboxylate(6)methyl thiazole-2-carboxylate(6)methyl thiazole-4-carboxylate(6)N-(thiazol-2-yl)acetamide(8)N-(thiazol-2-yl)benzamide(9)N-phenylthiazol-2-amine(9)tert-butyl thiazol-2-ylcarbamate(22)thiazol-2-ylmethanamine(5)thiazol-2-ylmethanol(9)thiazol-5-amine(6)thiazole-2-carbaldehyde(8)thiazole-2-carboxylic acid(9)thiazole-4-carboxylic acid(5)thiazole-5-carbaldehyde(13)thiazole-5-carbonitrile(7)thiazole-5-carboxylic acid(10)thiazolo[4,5-b]pyridine(12)thiazolo[5,4-b]pyridine(13)thiazolo[5,4-c]pyridine(5)thiazolo[5,4-d]pyrimidine(5)acetylthiazole(7)aminothiazole(24)bromothiazole(32)bromothiazolo(9)chlorothiazole(32)chlorothiazolo(18)cyanothiazole(7)cyclopropylthiazole(11)dibromothiazole(7)dichlorothiazole(8)dihydrothiazole(16)dihydrothiazolo(14)dimethylthiazole(22)dioxaborolane-thiazole(1)ethylthiazole(8)formylthiazole(7)imidazothiazole(42)methoxythiazole(43)methylthiazole(109)methylthiazolo(4)nitrothiazol(8)phenylthiazole(28)phenylthiazolidine(36)pyridine-thiazol-amide(23)pyridine-thiazole(135)tetrahydrothiazolo(8)thiazol-amide-hydrochloride(18)thiazol-ethanamine(4)thiazole-carboxylic acid(114)thiazole-hydrochloride(18)thiazole-nitrile(52)thiazolopyridine(68)thiazopyrimidine(50)trifluoromethyl-phenyl-thiazole(3)trifluoromethyl-thiazol(72)
Thiophenes(1488)
(9H-fluoren-9-yl)methyl (2-(thiophen-2-yl)ethyl)carbamate(5)1-(thiophen-2-yl)ethan-1-one(17)2,2'-bithiophene(25)2,2':5',2''-terthiophene(9)2,3-dihydrothieno[3,4-b][1,4]dioxine(9)2,5-dibromothiophene(18)2,5-diphenylthiophene(5)2-(thiophen-2-yl)pyridine(5)2-(trifluoromethyl)thiophene(11)2-chlorothiophene(20)2-ethynylthiophene(8)2-iodothiophene(14)2-methoxythiophene(7)2-nitrothiophene(20)3-bromothiophene(6)3-chlorothiophene(17)3-fluorothiophene(6)3-methoxythiophene(7)3-methylthiophene(14)3-nitrothiophene(6)3-phenylthiophene(13)4,4,5,5-tetramethyl-2-(thiophen-2-yl)-1,3,2-dioxaborolane(23)4,4,5,5-tetramethyl-2-(thiophen-3-yl)-1,3,2-dioxaborolane(10)4,5,6,7-tetrahydrothieno[2,3-c]pyridine(10)4,5,6,7-tetrahydrothieno[3,2-c]pyridine(9)4,7-di(thiophen-2-yl)benzo[c][1,2,5]thiadiazole(9)4,8-di(thiophen-2-yl)benzo[1,2-b:4,5-b']dithiophene(8)4-phenyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepine(5)4H-cyclopenta[2,1-b:3,4-b']dithiophene(9)4H-thieno[3,2-b]pyrrole(6)4H-thieno[3,4-c]pyrrole-4,6(5H)-dione(7)5,6-dihydro-4H-cyclopenta[b]thiophene(7)5-(thiophen-2-yl)isoxazole(5)5-benzyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine(7)5-phenylthiophen-2-amine(5)benzo[1,2-b:4,5-b']dithiophene(7)dibenzo[b,d]thiophene(22)dihydrothiophen-3(2H)-one(4)dithieno[3,2-b:2',3'-d]thiophene(5)ethyl thiophene-2-carboxylate(19)ethyl thiophene-3-carboxylate(11)methyl thiophene-3-carboxylate(33)N,N-diphenyl-4-(thiophen-2-yl)aniline(5)phenyl(thiophen-2-yl)methanone(6)thieno[2,3-b]pyridine(22)thieno[2,3-c]pyridine(14)thieno[2,3-d]pyrimidine(30)thieno[3,2-b]pyridine(18)thieno[3,2-b]thiophene(19)thieno[3,2-c]pyridine(13)thieno[3,2-d]pyrimidine(24)thieno[3,4-b]thiophene(5)thiophen-2-ylboronic acid(20)thiophen-2-ylmethanol(10)thiophen-3-amine(18)thiophen-3-ol(6)thiophene-2-carbaldehyde(38)thiophene-2-carbonitrile(23)thiophene-3-carbaldehyde(12)thiophene-3-carbonitrile(13)thiophene-3-carboxamide(5)thiophene-3-carboxylic acid(8)acetylthiophene(5)aminothiophene(23)bromothieno(29)chlorothieno(41)chlorothiophene(61)cyanothiophene(5)dibromothieno(5)dibromothiophene(17)dichlorothieno(10)dichlorothiophene(13)dihydrothieno(24)dihydrothiophene(1)dihydroxythiophene(7)dimethylthieno(10)dimethylthiophene(21)dithiophene(40)fluorothiophene(18)formylthiophene(16)hydroxythiophene(7)iodothieno(6)iodothiophene(13)methylthieno(27)nitrothiophene(27)oxotetrahydrothiophene(1)terthiophene(10)tetrahydrothieno(17)thiophen-amine-hydrochloride(52)thiophenecarboxaldehyde(10)thiphen-amine-hydrochloride(32)trithiophene(5)
Triazines(1392)
1,2,4-triazine(10)1,3,5-triazin-2-amine(3)2,4,6-triphenoxy-1,3,5-triazine(4)2,4,6-triphenyl-1,3,5-triazine(24)2,4-diphenyl-1,3,5-triazine(6)2-chloro-1,3,5-triazine(10)2-methoxy-1,3,5-triazine(10)2-phenyl-1,3,5-triazine(6)N2,N4,N6-triphenyl-1,3,5-triazine-2,4,6-triamine(4)pyrrolo[2,1-f][1,2,4]triazine(14)amino-triazine(31)benzotriazine(7)methyl-triazine(3)phenyl-triazine(4)pyrazolotriazine(4)triazin-amine(60)triazine-one(27)
Triazoles(7731)
(5aS,10bR)-2-phenyl-2,5a,6,10b-tetrahydro-4H-indeno[2,1-b][1,2,4]triazolo[4,3-d][1,4]oxazin-11-ium(6)1-(phenylsulfonyl)-1H-1,2,4-triazole(6)1-benzyl-1H-1,2,3-triazole(8)1-benzyl-1H-1,2,4-triazole(6)1-methyl-1H-1,2,3-triazole(9)1-methyl-1H-1,2,4-triazole(17)1-methyl-1H-benzo[d][1,2,3]triazole(10)1-phenethyl-1H-1,2,4-triazole(6)1-phenyl-1H-1,2,3-triazole(7)1-phenyl-1H-1,2,4-triazole(20)1H-1,2,4-triazol-3-amine(7)1H-1,2,4-triazol-5-amine(6)2-bromo-[1,2,4]triazolo[1,5-a]pyridine(6)2-phenyl-2,5,6,7-tetrahydropyrrolo[2,1-c][1,2,4]triazol-4-ium(6)2-phenyl-2H-1,2,3-triazole(7)2-phenyl-2H-benzo[d][1,2,3]triazole(8)2H-1,2,3-triazole(7)4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazole(7)4-phenyl-4H-1,2,4-triazole(6)4-phenyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepine(5)4H-1,2,4-triazole(13)5-bromo-1H-1,2,4-triazole(6)5-bromo-1H-benzo[d][1,2,3]triazole(5)5-fluoro-1H-benzo[d][1,2,3]triazole(4)6-bromo-[1,2,4]triazolo[1,5-a]pyridine(12)6-bromo-[1,2,4]triazolo[4,3-a]pyridine(8)8-bromo-[1,2,4]triazolo[1,5-a]pyridine(6)[1,2,4]triazolo[1,5-a]pyrazine(9)[1,2,4]triazolo[1,5-a]pyridin-2-amine(17)[1,2,4]triazolo[4,3-a]pyridin-3-ylmethanamine(5)ethyl 1H-1,2,4-triazole-3-carboxylate(3)amino-triazole(58)amino-triazolopyrimidine(4)benzotriazole(4)benzotriazole-ethylamine(2)bromo-triazole(143)bromotriazolopyridine(25)chloro-triazolopyridazine(3)chloro-triazolopyridine(29)chloro-triazolopyrimidine(6)fluoro-triazolopyridine(4)indenotriazolooxazine(5)methyl-triazole(61)methyl-triazolopyridine(5)methyl-triazolopyrimidine(16)nitro-triazole(2)phenyl-triazole(243)piperidine-triazole(20)triazol-amine(94)triazol-benzene(217)triazol-one(48)triazol-oxazine(5)triazol-pyridine(4)triazole-carbonitrile(5)triazole-carboxylate(37)triazole-carboxylic(5)triazole-carboxylic-acid(1)triazole-methanol(5)triazolopyrazine(49)triazolopyridazine(12)triazolopyridin-amine(41)triazolopyridine(51)triazolopyridine-carboxylate(16)triazolopyridine-carboxylic(6)triazolopyrimidine(41)
Organic Building Blocks
Alcohols(45662)
(1-methyl-1H-pyrazol-3-yl)methanol(7)(1-phenylcyclopropyl)methanol(3)(1H-benzo[d]imidazol-2-yl)methanol(13)(1H-indol-2-yl)methanol(7)(2-(trifluoromethyl)phenyl)methanol(6)(2-bromo-6-fluorophenyl)methanol(4)(2-bromophenyl)methanol(27)(2-chlorophenyl)methanol(34)(2-fluorophenyl)methanol(47)(2-iodophenyl)methanol(10)(2-nitrophenyl)methanol(17)(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol(7)(3-(trifluoromethoxy)phenyl)methanol(3)(3-(trifluoromethyl)phenyl)methanol(6)(3-chlorophenyl)methanol(21)(3-fluorophenyl)methanol(39)(3-iodophenyl)methanol(6)(3-methylpyridin-2-yl)methanol(10)(3-nitrophenyl)methanol(16)(3-phenoxyphenyl)methanol(3)(3-phenylisoxazol-5-yl)methanol(7)(4-fluorophenyl)methanol(18)(4-nitrophenyl)methanol(12)(6-methylpyridin-2-yl)methanol(5)(tetrahydrofuran-2-yl)methanol(6)1,2-diphenylethan-1-ol(8)1-phenyl-1H-pyrazol-3-ol(3)1-phenylethan-1-ol(69)2,2,2-trifluoro-1-phenylethan-1-ol(16)2-(1H-indol-3-yl)ethan-1-ol(10)2-(2-chlorophenyl)ethan-1-ol(6)2-(piperazin-1-yl)ethan-1-ol(9)2-(piperidin-1-yl)ethan-1-ol(10)2-(piperidin-2-yl)ethan-1-ol(8)2-(piperidin-4-yl)ethan-1-ol(5)2-amino-2-(2-fluorophenyl)ethan-1-ol(3)2-amino-2-(anthracen-9-yl)ethan-1-ol(4)2-amino-2-phenylethan-1-ol(96)2-hydroxy-1,2-diphenylethan-1-one(5)2-hydroxy-1-phenylethan-1-one(5)2-hydroxyethyl benzoate(3)2-phenoxyethan-1-ol(12)2-phenylethan-1-ol(47)2-phenylpropan-2-ol(11)3-phenylpropan-1-ol(18)4-fluoro-2-(hydroxymethyl)-5-(9H-purin-9-yl)tetrahydrofuran-3-ol(4)4-phenylpiperidin-4-ol(10)azetidin-2-ylmethanol(6)azetidin-3-ol(8)benzyl 3-(hydroxymethyl)piperazine-1-carboxylate(6)benzyl-2-(hydroxymethyl)pyrrolidine-1-carboxylate(9)bicyclo[1.1.1]pentan-1-ylmethanol(3)cyclobutanol(32)cyclobutylmethanol(9)cyclohexanol(50)cyclohexylmethanol(19)cyclopentanol(45)diphenylmethanol(18)isoxazol-3-ylmethanol(3)isoxazol-5-ylmethanol(5)m-tolylmethanol(15)morpholin-2-ylmethanol(12)morpholin-3-ylmethanol(14)naphthalen-1-ylmethanol(6)o-tolylmethanol(17)oxiran-2-ylmethanol(3)piperidin-2-ylmethanol(11)piperidin-3-ol(27)piperidin-3-ylmethanol(15)piperidin-4-ylmethanol(8)pyridin-2-ylmethanol(65)pyridin-3-ylmethanol(39)pyridin-4-ylmethanol(16)pyrimidin-2-ylmethanol(5)pyrrolidin-2-ylmethanol(26)pyrrolidin-3-ylmethanol(13)quinazolin-2-ol(7)tert-butyl 3-(hydroxymethyl)-1H-indole-1-carboxylate(10)thiazol-2-ylmethanol(9)thiophen-2-ylmethanol(10)triphenylmethanol(13)(2-hydroxyethoxy)carbonyl(3)(R)-1-ethanol(31)(R)-2-amino-ethanol(71)(R)-2-amino-propan-1-ol(5)(R)-2-hydroxy-acetic acid(7)(R)-2-hydroxypropanoic acid(2)(R)-ethane-1,2-diol(11)(R)-tert-butyl 2-hydroxyethyl-carbamate(3)(S)-1-ethanol(19)(S)-2-amino-ethanol(79)(S)-2-amino-propan-1-ol(7)(S)-2-hydroxy-acetic acid(8)(S)-3-amino-propan-1-ol(2)(S)-tert-butyl (3-hydroxypropan-2-yl)carbamate(6)(S)-tert-butyl 2-hydroxyethyl-carbamate(6)1,1,1,3,3,3-hexafluoropropan-2-ol(7)1-ethanol(84)2,2,2-trifluoro ethanol(24)2,2-dihydroxy-ethanone(5)2-amino-propan-1-ol(8)2-aminoethanol(34)2-hydroxyacetic acid(15)2-hydroxyacetonitrile(5)2-hydroxyethanone(9)2-hydroxyethyl(4)2-hydroxypropanesulfonic acid(3)2-hydroxypropanoic acid(5)2-methylpropan-1-ol(6)2-methylpropan-2-ol(5)3-amino-1-propanol(6)amino-2-methylpropan-2-ol(2)amino-alcohol-hydrochloride(78)amino-ethanol(25)amino-hydroxypropanamide-hydrochloride(2)amino-methanol(4)amino-methyl-butanol(7)amino-propanol(6)aminohydroxy(5)aminomethyl-ol(6)benzofuran-methanol(2)benzoimidazol-methanol(4)benzoxazole-methanol(4)benzylaminoethanol(3)bromo-hydroxy(327)butan-1-ol(13)carbazole-hydroxyl(5)cyclobutyl-methanol(4)cyclopropylethanol(6)diethanol(23)diethanol(23)dihydroxy(7)dihydroxy-amino(2)dihydroxy-benzene(65)dihydroxy-pyridine(14)dihydroxybenzylidene(4)dihydroxynaphthalene(7)dihydroxypentan(2)dihydroxypentan(2)dihydroxypropanoate(7)dihydroxypropyl-isophthalamide(6)dihydroxypyrimidine(5)dihydroxypyrrolidine(3)dihydroxyterephthalic(3)dihydroxyterephthalic acid(1)dimethanol(9)dioxaborolan-methanol(4)ethane-1,2-diol(8)ethanol(555)ethoxyethanol(29)ethyl-hydroxy(7)ethylaminoethanol(3)ethynyl-ol(7)fluorene-methanol(1)hydroxy-benzimidazole(23)hydroxy-benzofuran(11)hydroxy-benzothiazole(17)hydroxy-carboxylic acid(9)hydroxy-hydroxymethyl(5)hydroxy-indoline(3)hydroxy-naphthyridine(16)hydroxy-pyrazole(74)hydroxy-tetrahydroisoquinolin(3)hydroxy-trifluoromethyl(8)hydroxyacetamide(1)hydroxyacetate(7)hydroxyacetimidamide(3)hydroxyadamantan(4)hydroxyadamantan(4)hydroxyazetidine(7)hydroxybenzofuran(8)hydroxybenzyl(18)hydroxybenzylidene(5)hydroxybicyclo(7)hydroxybutan(4)hydroxybutan(4)hydroxybutanoate(36)hydroxybutanoic(29)hydroxybutyrate(24)hydroxycarbamimidoyl(2)hydroxycinnamic(3)hydroxycinnamic acid(2)hydroxycyclobutyl(3)hydroxycyclohexyl(7)hydroxycyclopentyl(1)hydroxydiphenylmethyl(2)hydroxyethoxy(22)hydroxyethoxy(22)hydroxyethyl-morpholine(3)hydroxyethyl-piperidine-carboxylate(3)hydroxyhexanoate(8)hydroxyhexanoic(6)hydroxyhexyl(8)hydroxyhexyl(8)hydroxyimino(11)hydroxyisobenzofuran(2)hydroxyisonicotinaldehyde(7)hydroxyisonicotinate(5)hydroxyisonicotinic(5)hydroxyisonicotinic(5)hydroxyisonicotinic acid(3)hydroxyisoquinoline(12)hydroxylethyl(3)hydroxymethy-indole-carboxylate(2)hydroxymethyl-phenyl-boronic acid(37)hydroxymethyl-phenyl-boronic-acid(12)hydroxymethyl-piperazine-carboxylate(17)hydroxymethyl-piperidine-carboxylate(23)hydroxymethyl-tetrahydropyran(3)hydroxynaphthalene(29)hydroxynicotinaldehyde(7)hydroxynicotinate(13)hydroxynicotinonitrile(6)hydroxyoctadec(1)hydroxyoctadec(1)hydroxyoctadecanoic(7)hydroxypentanedioate(1)hydroxypentanoate(6)hydroxypiperidine(46)hydroxypropanoic(35)hydroxypyridazine(5)hydroxypyridine(33)hydroxypyrimidine(19)hydroxypyrrolidine(96)hydroxyquinoline(68)imidazol-methanol(22)imidazolyl-ethanol(3)indazole-hydroxyl(10)indazole-methanol(6)indol-methanol(8)indole-ethanol(6)indole-hydroxyl(138)isoquinoline-methanol(7)methanol-hydrochloride(38)methyl 2-hydroxyacetate(5)methyl hydroxycarboxylate(9)methyl-butanediol(2)methyl-butanol(7)methyl-ol(52)methylmethanol(10)minodiethanol(11)morpholin-methanol(21)oxypropane-1,2-dio(7)pentahydroxyhexan(2)pentahydroxyhexan(2)pentahydroxyhexanal(9)phenyl-ethanol(69)phenyl-methanol(392)phenyl-propanol(12)phenylhydroxyl(22)piperazine-hydroxyl(73)piperazine-methanol(5)piperidine-ethanol(16)piperidinemethanol(6)prop-2-en-1-ol(6)prop-2-yn-1-ol(8)propan-1-ol(45)propan-2-ol(54)propane-1,2-diol(4)propane-1,3-diol(5)propanediol(8)propanediol(8)pyrazol-methanol(34)pyrazole-ethanol(11)pyridinemethanol(8)pyridyl-ethanol(49)pyrimidine-methanol(46)pyrrolidine-methanol(18)pyrrolopyridine-hydroxy(2)pyrrolopyridine-methanol(3)tert-butyl hydroxymethylcarbamate(3)tetrahydrofuran-methanol(5)tetrahydroisoquinoline-methanol(2)tetrahydroxy(25)tetrahydroxy(25)tetrahydroxyhexanal(9)tetrahydroxypentanal(5)thiadiazole-methanol(3)thioethanol(5)triazole-methanol(5)trifluoromethyl-phenyl-ethanol(22)trifluoromethyl-phenyl-methanol(29)trifluoromethyl-pyridin-methanol(12)trihydroxy(246)trihydroxy(246)
Aldehydes(18770)
1-methyl-1H-indole-3-carbaldehyde(9)1-naphthaldehyde(23)1-phenyl-1H-pyrazole-4-carbaldehyde(4)1H-benzo[d]imidazole-2-carbaldehyde(4)1H-indazole-3-carbaldehyde(19)1H-indole-2-carbaldehyde(18)1H-pyrazole-4-carbaldehyde(17)1H-pyrrole-2-carbaldehyde(17)1H-pyrrole-3-carbaldehyde(7)1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde(7)1H-pyrrolo[2,3-b]pyridine-5-carbaldehyde(5)2,3-dichlorobenzaldehyde(6)2,3-difluorobenzaldehyde(9)2,4-dichlorobenzaldehyde(6)2,4-difluorobenzaldehyde(14)2,6-dibromobenzaldehyde(4)2,6-difluorobenzaldehyde(16)2-(benzyloxy)benzaldehyde(13)2-(phenylethynyl)benzaldehyde(6)2-(trifluoromethoxy)benzaldehyde(5)2-(trifluoromethyl)benzaldehyde(16)2-bromo-3-hydroxybenzaldehyde(4)2-bromo-3-methoxybenzaldehyde(4)2-bromo-6-fluorobenzaldehyde(11)2-chloro-3-hydroxybenzaldehyde(4)2-chloro-3-methoxybenzaldehyde(4)2-chloro-4-fluorobenzaldehyde(8)2-fluoro-3-methylbenzaldehyde(7)2-fluorobenzaldehyde(75)2-hydroxy-3-iodobenzaldehyde(3)2-iodobenzaldehyde(16)2-methyl-1H-indole-3-carbaldehyde(3)2-naphthaldehyde(11)2-nitrobenzaldehyde(33)2-oxo-2-phenylacetaldehyde(7)2-phenylacetaldehyde(17)3-(benzyloxy)benzaldehyde(12)3-(trifluoromethoxy)benzaldehyde(5)3-(trifluoromethyl)benzaldehyde(20)3-bromo-2-chlorobenzaldehyde(4)3-bromo-2-fluorobenzaldehyde(9)3-bromo-2-hydroxybenzaldehyde(7)3-bromo-4-fluorobenzaldehyde(7)3-bromo-4-hydroxybenzaldehyde(5)3-bromo-4-methoxybenzaldehyde(7)3-chloro-2-fluorobenzaldehyde(8)3-chloro-2-hydroxybenzaldehyde(4)3-chloro-4-methoxybenzaldehyde(3)3-chlorobenzaldehyde(53)3-fluorobenzaldehyde(58)3-iodobenzaldehyde(17)3-methoxy-2-nitrobenzaldehyde(3)3-nitrobenzaldehyde(28)3-phenoxybenzaldehyde(4)4-(1H-imidazol-1-yl)benzaldehyde(6)4-(benzyloxy)benzaldehyde(15)4-(piperazin-1-yl)benzaldehyde(5)4-fluorobenzaldehyde(44)4-hydroxy-3-nitrobenzaldehyde(5)4-morpholinobenzaldehyde(8)4-nitrobenzaldehyde(11)4-phenoxybenzaldehyde(7)5-phenylfuran-2-carbaldehyde(9)6-methoxynicotinaldehyde(4)[1,1'-biphenyl]-3-carbaldehyde(11)[1,1'-biphenyl]-4-carbaldehyde(27)benzo[b]thiophene-2-carbaldehyde(6)benzo[b]thiophene-3-carbaldehyde(6)benzo[d][1,3]dioxole-5-carbaldehyde(4)benzofuran-2-carbaldehyde(5)furan-2-carbaldehyde(19)imidazo[1,2-a]pyridine-2-carbaldehyde(8)imidazo[1,2-a]pyridine-3-carbaldehyde(8)isonicotinaldehyde(35)methyl 3-formylbenzoate(7)nicotinaldehyde(107)picolinaldehyde(82)pyrazine-2-carbaldehyde(15)pyrimidine-5-carbaldehyde(21)thiazole-2-carbaldehyde(8)thiazole-5-carbaldehyde(13)thiophene-2-carbaldehyde(38)thiophene-3-carbaldehyde(12)2-oxoacetaldehyde(11)acetaldehyde(28)aminonicotinaldehyde(5)aminonicotinaldehyde(5)benzenedicarboxaldehyde(1)carboxyaldehyde(1094)dicarbaldehyde(37)dicarbaldehyde(37)formyl-indole-carboxylate(15)formyl-pyrrole-carboxylate(14)formylisonicotinate(7)nitropicolinaldehyde(3)phenylpropanal(8)propionaldehyde(10)tetracarbaldehyde(7)tetracarbaldehyde(7)tricarbaldehyde(10)
Aliphatic Chain Hydrocarbons(11899)
acetylthio(7)acetylthio(7)azanonadecan(7)azanylylidene(1)azapentadecan(10)azapentadecan(10)azatridecan(4)butyloctyl(4)butyloctyl(4)carbonylbis(2)carbonylbis(2)diazirine(13)diazirine(13)didodecyl(7)didodecyl(7)diethyl(73)diethyl(73)dihydroxypropan(6)diisobutyl(2)diisopropyl(27)diisopropyl(27)dimethoxy(306)dimethyladamantane(14)dimethyladamantane(14)dimethylalkanes(7)dinonyl(5)dinonyl(5)dioctyl(1)dioxide(451)dioxide(451)ethoxy(712)ethoxyethoxy(5)ethoxyethoxy(5)ethoxyethyl(16)ethoxyethyl(16)ethoxymethyl(11)ethoxymethylene(1)ethoxymethylene(1)hemihydrate(11)hemihydrate(11)hexahydrate(38)hexahydrate(38)isobutyryl(2)isobutyryl(2)isopropylidenebis(2)isopropylsulfonyl(10)isopropylsulfonyl(10)methoxybut(2)methoxybut(2)methoxyethoxy(67)methoxyethoxy(67)methoxyethyl(69)methoxyisonicotinic(11)methoxyisonicotinic(11)methoxymethoxy(23)methoxymethoxy(23)methoxymethyl(117)methoxynicotinic(26)methoxynicotinic(26)methoxynicotinic acid(11)methoxypropan(23)methoxypropan(23)methoxypropoxy(8)methoxypropoxy(8)methoxypropyl(10)methoxytetrahydro(14)methoxytetrahydro(14)methyladamantan(4)methylisonicotinic(13)oxoethoxy(1)oxopropane(10)oxopropyl(32)oxopropyl(32)pentahydrate(7)pentahydrate(7)pentyloxy(7)tetrahydrate(18)tetrahydrate(18)tetramethoxy(6)triisopropyl(32)triisopropyl(32)trimethoxy(15)trimethoxy(15)trioxatriborinane(5)yldisulfanyl(7)
Aliphatic Cyclic Hydrocarbons(9963)
(1-phenylcyclobutyl)methanamine(9)(1-phenylcyclopropyl)methanamine(11)(1-phenylcyclopropyl)methanol(3)(3aR,E)-4-((Z)-2-(2-methylenecyclohexylidene)ethylidene)octahydro-1H-indene(7)(4-cyclohexylphenyl)boronic acid(7)(4-pentylcyclohexyl)benzene(5)(4-propylcyclohexyl)benzene(5)(9H-fluoren-2-yl)boronic acid(5)(9H-fluoren-9-yl)methyl (4-methoxybenzyl)carbamate(7)(cyclopropylmethoxy)benzene(18)1,1-difluorocyclobutane(14)1,1-difluorocyclohexane(16)1,1-difluorocyclopentane(5)1-bromo-2-cyclopropylbenzene(3)1-bromo-3-cyclopropylbenzene(6)1-chloro-2-cyclopropylbenzene(7)1-cyclohexyl-2-fluorobenzene(4)1-cyclopropyl-3-methylbenzene(3)1-methylcyclohex-1-ene(25)1-phenylcyclobutan-1-amine(5)1-phenylcyclobutane-1-carbonitrile(6)1-phenylcyclobutane-1-carboxylic acid(16)1-phenylcyclohexane-1-carboxylic acid(5)1-phenylcyclopropan-1-amine(26)1-phenylcyclopropane-1-carbonitrile(19)1-phenylcyclopropane-1-carboxylic acid(20)2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-2-phenylacetic acid(5)2-bromo-9,9-diphenyl-9H-fluorene(5)2-bromo-9H-fluorene(15)2-cyclohexylacetic acid(12)2-cyclopropyl-4-phenylquinoline(12)2-cyclopropylpyridine(20)2-cyclopropylpyrimidine(10)2-iodo-9H-fluorene(8)2-methylcyclohexan-1-one(2)2-phenylcyclopropan-1-amine(20)3,3-dimethylcyclohexan-1-one(4)3-cyclopropylpyridine(28)3-methylcyclohexan-1-one(7)4-cyclohexylbenzoic acid(6)4-cyclohexylbenzonitrile(10)4-cyclohexylphenol(7)5-azaspiro[2.4]heptane(13)6-azaspiro[2.5]octane(10)9,9-dimethyl-9H-fluorene(16)bicyclo[1.1.1]pentan-1-ylmethanol(3)bicyclo[1.1.1]pentane-1-carboxylic acid(6)bicyclo[2.2.1]hept-2-ene(18)bicyclo[2.2.1]heptane(15)bromocyclohexane(6)cyclobutanamine(90)cyclobutanecarboxylic acid(23)cyclobutanol(32)cyclobutylmethanol(9)cycloheptane(28)cyclohex-3-ene-1-carboxylic acid(7)cyclohexanamine(45)cyclohexanecarboxylic acid(54)cyclohexanol(50)cyclohexylmethanol(19)cyclopentanamine(5)cyclopentanecarboxylic acid(26)cyclopentanol(45)cyclopropoxybenzene(15)ethyl 1-phenylcyclopropane-1-carboxylate(3)ethyl 2-phenylcyclopropane-1-carboxylate(5)ethyl cyclohexanecarboxylate(19)isopropylcyclohexane(18)methoxycyclohexane(5)methyl 1-phenylcyclopropane-1-carboxylate(10)methyl 4-oxocyclohexane-1-carboxylate(4)methyl bicyclo[1.1.1]pentane-1-carboxylate(9)methyl cyclobutanecarboxylate(19)methyl cyclohexanecarboxylate(26)methyl cyclopentanecarboxylate(16)N-methylcyclohexanamine(7)naphthalene-1,4-dione(23)spiro[3.3]heptane(11)tert-butyl (1-phenylcyclopropyl)carbamate(6)tert-butyl (2-phenylcyclopropyl)carbamate(3)tert-butyl bicyclo[1.1.1]pentan-1-ylcarbamate(5)tert-butyl cyclobutylcarbamate(17)tert-butyl cyclohexylcarbamate(9)tert-butyl cyclopentylcarbamate(28)adamantan(36)aminobicyclo(24)aminocyclobutanol(5)aminocyclobutyl(5)aminocyclohexane(55)aminocyclohexanol(21)aminocyclopentanol(17)aminocyclopentyl(6)aminocyclopropyl(4)azabicyclo(292)azabicyclo(292)azatricyclo(5)azatricyclo(5)azetidine(185)azetidine(185)benzylazetidin(4)bicyclo(61)bicyclohexyl(6)bromobicyclo(9)butylcyclohexyl(9)butylsulfonyl(6)chlorocyclopropyl(2)cyanoazetidine(5)cyanocyclopropyl(4)cyclobutane(42)cyclobutanecarbonitrile(2)cyclobutanecarboxylate(7)cyclobutanecarboxylic(9)cyclobutyl-hydrochloride(16)cyclobutyl-methanol(4)cyclobutylacetic(4)cyclobutylmethyl(4)cyclobutylmethyl(4)cyclodextrin(453)cyclodextrin(453)cyclohexa(176)cyclohexa(176)cyclohexane(127)cyclohexane(127)cyclohexanecarbonitrile(23)cyclohexanecarboxylate(5)cyclohexanecarboxylic(19)cyclohexanediamine(8)cyclohexanedicarboxylic(3)cyclohexanone(59)cyclohexylacetic(2)cyclohexylamino(9)cyclohexylbutanoic(4)cyclohexylcarbamoyl(5)cyclohexylethanamine(12)cyclohexylmethoxy(4)cyclohexylmethoxy(4)cyclohexylmethyl(7)cyclohexylmethyl(7)cyclohexyloxy(10)cyclohexylpropan(94)cyclohexylpropanoic(9)cyclopentadienyl(2)cyclopentane(169)cyclopentane(169)cyclopentanecarboxylate(3)cyclopentanecarboxylic(7)cyclopentyl(56)cyclopentyl(56)cyclopentylacetate(14)cyclopentylacetic(3)cyclopentylamino(1)cyclopentylethanamine(2)cyclopentylethanamine(2)cyclopentylmethyl(4)cyclopentyloxy(16)cyclopentyloxy(16)cyclopentylpropan(3)cyclopentylpropanoic(1)cyclopropanamine(32)cyclopropanamine(32)cyclopropanecarbonitrile(14)cyclopropanecarbonyl(6)cyclopropanecarbonyl(6)cyclopropanecarboxamide(14)cyclopropanecarboxylate(11)cyclopropanecarboxylic(23)cyclopropanol(4)cyclopropoxyphenyl(7)cyclopropyl(356)cyclopropyl(356)cyclopropylacetate(6)cyclopropylacetic(3)cyclopropylamino(24)cyclopropylcarbamoyl(5)cyclopropylethan(5)cyclopropylethan(5)cyclopropylethanone(2)cyclopropylethynyl(1)cyclopropylhydrazine(4)cyclopropylhydrazine(4)cyclopropylmethoxy(32)cyclopropylmethoxy(32)cyclopropylmethyl(22)cyclopropylmethyl(22)cyclopropylnicotinamide(4)cyclopropylpiperidin(1)cyclopropylpropanoic(5)cyclopropylpyrimidine(18)cyclopropylsulfonyl(6)cyclopropylsulfonyl(6)diazabicyclo(77)diazabicyclo(77)dicyclohexyl(6)dicyclohexyl(6)dicyclohexylphosphino(18)dicyclohexylphosphino(18)difluorocyclobutyl(6)difluorocyclohexyl(7)dihydroinden(8)dihydroindeno(5)dihydroindeno(5)dihydrophenazine(3)dihydrophenazine(3)dihydropteridin(6)dimethylazetidin(16)dimethylazetidin(16)dimethylbicyclo(5)dimethylbicyclo(5)dimethylcyclohex(5)dimethylcyclohexane(16)dimethylcyclohexanone(7)dimethylcyclopropanecarboxylic(8)dioxathiane(4)dioxathiolane(1)dioxathiolane(1)dioxol(175)dithiolan(1)dithiolan(1)ethylcyclohexanecarboxylate(2)ethylcyclohexyl(4)ethylcyclohexyl(4)ethylsulfonyl(42)fluoroazetidine(1)fluorobicyclo(4)hexahydrocyclohepta(6)hexahydrocyclohepta(6)hexahydrocyclopenta(4)hydroxycyclobutanecarboxylate(4)hydroxycyclobutanecarboxylic(4)hydroxycyclohexanecarboxylate(10)hydroxycyclohexanecarboxylic(6)hydroxycyclopentanecarboxylate(4)methanesulfonyl(314)methanesulfonyl(314)methoxycyclohexanamine(6)methylazetidine(30)methylbicyclo(6)methylbicyclo(6)methylcyclobutanamine(5)methylcyclobutanecarboxylate(5)methylcyclobutanecarboxylic(5)methylcyclohexanamine(11)methylcyclohexane(8)methylcyclohexane(8)methylcyclohexanecarboxylic(5)methylcyclohexanecarboxylic acid(3)methylcyclohexanol(21)methylcyclohexanone(7)methylcyclohexyl(33)methylcyclopentane(2)methylcyclopropanamine(4)methylcyclopropanamine(4)methylcyclopropane(16)methylsulfinyl(24)methylsulfonimidoyl(6)methylsulfonimidoyl(6)methylsulphonyl(10)octahydrocyclopenta(14)oxabicyclo(40)oxabicyclo(40)oxazepane(13)oxetane(42)oxetane(42)Oxetanes(143)oxoazetidine(8)oxoazetidine(8)oxobicyclo(10)oxobicyclo(10)oxocyclobutanecarboxylate(3)oxocyclobutyl(2)oxocyclohex(3)oxocyclohexane(4)oxocyclohexane(4)oxocyclohexanecarbonitrile(3)oxocyclohexanecarboxylate(11)oxocyclohexanecarboxylic(6)oxocyclohexyl(3)oxocyclohexyl(3)oxocyclopentane(2)oxocyclopentanecarboxylate(5)oxocyclopentyl(5)pentamethylcyclopentadienyl(11)pentylcyclohexyl(10)pentylcyclohexyl(10)phenylcyclopropanamine(8)propylcyclohexyl(15)propylcyclohexyl(15)spiro-amine-hydrochloride(1)tetraazacyclododecane(13)tetraazacyclododecane(13)tetraazacyclotetradecane(7)tetraazacyclotetradecane(7)tetramethylcyclohexane(4)tetraoxatridecan(3)tetraoxatridecan(3)thiazepine(2)tricyclohexylphosphine(9)trimethylbicyclo(21)trimethylcyclohex(20)trimethylcyclohexane(4)trimethylcyclohexane(4)
Alkenyls(16041)
(1E,4E)-1,5-diphenylpenta-1,4-dien-3-one(7)(1E,6E)-1,7-diphenylhepta-1,6-diene-3,5-dione(7)(2,2-dibromovinyl)benzene(6)(2-(4-bromophenyl)ethene-1,1,2-triyl)tribenzene(11)(2-nitrovinyl)benzene(16)(allyloxy)benzene(15)(E)-5-benzylidenethiazolidine-2,4-dione(6)(E)-[3,3'-biindolinylidene]-2,2'-dione(5)(E)-N-(3-oxo-1-phenyl-3-(piperidin-1-yl)prop-1-en-2-yl)benzamide(6)(Z)-3-(phenyl(phenylamino)methylene)indolin-2-one(5)(Z)-3-benzylideneindolin-2-one(7)1,2-diphenylethene(5)1,3,3-trimethylcyclohex-1-ene(12)1-bromo-4-styrylbenzene(5)1-methylcyclohex-1-ene(25)2-Benzalacetophenone(30)2-methoxycyclohexa-2,5-diene-1,4-dione(7)2-methylcyclohexa-2,5-diene-1,4-dione(5)3-(2-fluorophenyl)acrylic acid(5)3-(3-chlorophenyl)acrylic acid(6)3-methylene-2,3-dihydro-1H-inden-1-one(5)3-methyleneazetidine(8)3-phenylacrylaldehyde(22)3-styrylphenol(11)4-(1,2,2-triphenylvinyl)benzaldehyde(5)4-(prop-1-en-2-yl)cyclohex-1-ene(5)5H-dibenzo[b,f]azepine(5)allyl benzoate(4)cyclohex-1-en-1-yl trifluoromethanesulfonate(3)cyclohex-1-en-1-ylboronic acid(4)cyclohex-1-ene-1-carboxylic acid(3)cyclohexa-1,3-diene(5)cyclopent-2-en-1-amine(4)ethyl cyclohex-1-ene-1-carboxylate(5)methyl cinnamate(21)methyl cyclohex-3-ene-1-carboxylate(2)methylenecyclobutane(8)methylenecyclohexane(8)N-phenylacrylamide(5)styrene(56)tert-butyl-cyclopent-2-en-1-ylcarbamate(6)vinylcyclopropane(7)(dimethylamino)but-2-enamide(6)(E)-(2-nitrovinyl)(23)2,2-dibromovinyl(6)3-buten(7)3-buten-2-one(10)4-oxobut-2-enoic acid(4)6-oxy-hexyl acrylate(5)acrylaldehyde(11)acrylamide(52)acrylic acid(131)acryloyl(9)acryloyl(9)acryloyl chloride(5)alkenyl-phenyl-boronic-acid(2)allyl(54)allyl carboxylate(8)allyloxy(28)annulene(2)but-3-en-1-amine(6)butenoate(3)cinnamic acid(5)cyclohexene(67)cyclooctene(9)cyclopentene(3)diacrylamide(6)diallyl(5)diazene(21)divinyl(1)divinyl(1)enamide(6)enamide(6)enamine-hydrochloride(2)enecarbaldehyde(2)enoyl(9)ethenyl(143)ethoxyvinyl(48)ethyl 3-acrylate(30)ethyl acrylate(5)methacrylamide(3)methacrylamide(3)methoxycinnamaldehyde(3)methyl methacrylate(5)nitroethylene(37)prop-1-en(8)prop-1-en-2-yl(27)prop-2-en-1-ol(6)prop-2-en-1-one(13)tetrahydrocyclopenta(19)trienone(4)vinylboronic acid(10)
Alkyls(185)
(S)-sec-butyl(12)2,4,4-trimethylpentan(4)2-ethylhexyl(21)4,4-bis(2-ethylhexyl)(3)butyl(120)butyl-ium(12)decyl(19)dihexyl(5)dodecyl(25)ethyl(671)ethyl-methyl(5)ethylhexyl(37)heptadecan-9-yl(6)heptyl(15)hexadecy(5)hexyl(55)hexyldecyl(4)Isobutyl(89)isopropyl(610)isopropyl-phenyl-boronic acid(37)isopropyl-phenyl-boronic acid/ester(9)isopropyl-phenyl-boronic-acid(1)methylpentan(44)N-butyl-N-methyl(4)octyl(47)octyldodecyl(7)pentan-3-yl(5)pentyl(83)propyl(214)sec-butyl(9)tert-pentyl(9)tertbutyl(592)tetradecyl(6)undecyl(3)
Alkynyls(2694)
(bromoethynyl)benzene(5)(prop-2-yn-1-yloxy)benzene(8)1,3,5-tris(phenylethynyl)benzene(6)1,4-bis(phenylethynyl)benzene(7)1,4-diphenylbuta-1,3-diyne(8)1-ethynyl-2-fluorobenzene(6)1-methyl-4-(phenylethynyl)benzene(6)2-(phenylethynyl)benzaldehyde(6)2-ethynylpyridine(29)2-ethynylthiophene(8)3-ethynylpyridine(19)4-(phenylethynyl)-1,1'-biphenyl(5)4-(phenylethynyl)phenol(7)4-ethynylpyridine(8)9,10-bis(phenylethynyl)anthracene(5)ethynylbenzene(81)trimethyl(phenylethynyl)silane(21)(trimethylsilyl)ethynyl(36)2-propyn-1-ylcarbamoyl(5)diethynyl(7)diethynylbenzene(6)ethynyl(257)ethynyl-methylbenzene(10)ethynyl-nitrobenzene(9)ethynyl-ol(7)ethynylpyridine(23)N-(2-propynyl)-1-amine(6)phenylethene(17)prop-1-yn-1-yl(10)prop-2-yn-1-ol(8)prop-2-yn-1-oxy(25)prop-2-yn-1-yl(18)propargyl(5)propargyloxy(2)propiolate(5)propiolic(6)propiolic acid(11)propyn(3)propynyloxy(6)triisopropylsilylethynyl(8)
Amides(89900)
(3-(phenylcarbamoyl)phenyl)boronic acid(3)(4-(phenylcarbamoyl)phenyl)boronic acid(9)(9H-fluoren-9-yl)methyl (3-phenylpropyl)carbamate(24)(S)-N-phenethyl-3-(pyrrolidin-2-yl)propanamide(5)(S)-N-phenylpiperidine-2-carboxamide(1)1-(pyridin-2-yl-l2-azaneyl)ethan-1-one(12)1-methylindoline-2,3-dione(8)1-phenylurea(7)1H-pyrazole-5-carboxamide(3)2,2-dimethyl-1-(pyridin-2-yl-l2-azaneyl)propan-1-one(7)2-((tert-butoxycarbonyl)amino)benzoic acid(8)2-(trifluoromethyl)benzamide(4)2-amino-3-phenylpropanamide(7)2-amino-N-phenylbenzamide(20)2-aminobenzamide(17)2-bromo-N-phenylacetamide(5)2-chloro-N-phenylacetamide(21)2-chloro-N-phenylbenzamide(4)2-chlorobenzamide(12)2-fluoro-N-methylbenzamide(8)2-fluorobenzamide(22)2-hydroxy-N-phenylbenzamide(3)2-hydroxybenzamide(11)2-iodobenzamide(4)2-methoxybenzamide(5)3-benzamidopropanoic acid(3)3-bromo-4-fluorobenzamide(3)3-fluorobenzamide(15)3-nitrobenzamide(11)3-oxo-N-phenylbutanamide(8)4-amino-N-phenylbenzamide(7)4-benzamidobenzoic acid(3)4-fluorobenzamide(10)4-oxo-4-(phenylamino)butanoic acid(6)benzoylglycine(13)benzyl benzylcarbamate(16)benzyl cyclohexylcarbamate(9)benzyl phenylcarbamate(15)ethyl 2-oxo-2-(phenylamino)acetate(6)isonicotinamide(12)N,N-diethylbenzamide(9)N,N-dimethylbenzamide(16)N-((5R,6R)-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-6-yl)-3-phenylisoxazole-4-carboxamide(1)N-((6R,7R)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-7-yl)-2-phenylacetamide(5)N-(2-(1H-indol-3-yl)ethyl)acetamide(5)N-(2-benzoylphenyl)-2-bromoacetamide(3)N-(2-chlorophenyl)acetamide(13)N-(2-iodophenyl)acetamide(9)N-(2-nitrophenyl)acetamide(6)N-(3-fluorophenyl)acetamide(6)N-(4-aminophenyl)benzamide(2)N-(4-chlorophenyl)acetamide(6)N-(4-chlorophenyl)benzamide(6)N-(4-fluorophenyl)benzamide(3)N-(4-phenoxyphenyl)acetamide(4)N-(o-tolyl)benzamide(9)N-(p-tolyl)benzamide(6)N-(thiazol-2-yl)acetamide(8)N-isopropylbenzamide(10)N-methoxy-N-methylbenzamide(14)N-methylbenzamide(30)N-methylpicolinamide(10)N-phenylacrylamide(5)N-phenylpivalamide(8)nicotinamide(64)phenyl dimethylcarbamate(5)phenyl ethyl(methyl)carbamate(5)picolinamide(38)piperidine-3-carboxamide(5)pyrazine-2-carboxamide(10)pyrrolidine-2-carboxamide(12)tert-butyl (1-phenylethyl)carbamate(14)tert-butyl (4-fluorophenyl)carbamate(5)tert-butyl benzylcarbamate(42)tert-butyl methyl(phenyl)carbamate(8)tert-butyl o-tolylcarbamate(7)tert-butyl phenethylcarbamate(9)tert-butyl-cyclopent-2-en-1-ylcarbamate(6)thiophene-3-carboxamide(5)(S)-2-amino-propanamide(7)(S)-methyl 3-amino-propanoate(19)2,2,2-trifluoroacetamido(5)2-acetamide(34)2-acetamido-propanoic acid(5)2-amino-acetamide(7)2-bromo-acetamide(11)2-chloro-N-acetamide(44)2-chloro-N-methyl-acetamide(4)2-chloro-N-methylacetamide(4)2-propyn-1-ylcarbamoyl(5)3-chloro-propanamide(4)3-oxobutanamide(8)4-amino-4-oxobutanoic acid(13)acetamide(651)acetamide(651)acetamido(156)acetamidomethyl(11)acrylamide(52)amino-2-oxoacetic acid(7)amino-benzamide(4)amino-hydroxypropanamide-hydrochloride(2)benzamide(368)benzothioamide(40)bromo-picolin-amide(9)butyrylamino(5)carboxamide(929)carboxamide(929)diacetamide(3)dicarboxamide(19)diethylacetamide(4)diethylacetamide(4)diethylaminoethyl-carboxamide(8)diethylcarbamoyl(16)dihydroxypropyl-isophthalamide(6)diisopropylcarbamoyl(6)dimethylacetamide(9)dimethylnicotinamide(7)dimethylnicotinamide(7)dipropylcarbamoyl(2)ethyl 2-(amino)-2-oxoacetate(5)formamide(15)formamidophenylboronic acid(14)formamidophenylboronic acid/ester(3)isobutyramide(7)maleimide(2)maleimide(2)mercaptopropanamido(4)methoxybenzamide(26)N,N-dimethyl-acetamide(5)N,N-dimethylamide(61)N-butyl-carboxamide(3)N-ethyl-carboxamide(14)N-ethylacetamide(10)N-hydroxy-carboxamide(16)N-isopropyl-carboxamide(11)N-methoxy-N-methylamide(38)N-methyl-carboxamide(105)N-methylacetamide(8)oxobutanamide(10)oxobutanamide(10)pentanamide(18)pentanamide(18)pivalamide(28)propanamide(47)propanamido(5)propanamido(5)propionamido(6)tert-butylcarbamoy(13)thioamide(33)
Amines(139484)
(1-phenylcyclobutyl)methanamine(9)(1-phenylcyclopropyl)methanamine(11)(1H-benzo[d]imidazol-2-yl)methanamine(6)(2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methanamine(3)(2-aminophenyl)(phenyl)methanone(22)(2-bromophenyl)methanamine(11)(2-chlorophenyl)methanamine(20)(2-fluorophenyl)methanamine(30)(3-chlorophenyl)methanamine(12)(3-fluoropyridin-2-yl)methanamine(5)(3-methylpyridin-2-yl)methanamine(5)(4-aminophenyl)(phenyl)methanone(2)(4-fluorophenyl)methanamine(17)(6-methylpyridin-2-yl)methanamine(6)(tetrahydrofuran-2-yl)methanamine(4)(tetrahydrofuran-3-yl)methanamine(3)1,2,3,4-tetrahydroisoquinolin-5-amine(4)1,2,3,4-tetrahydroisoquinolin-6-amine(3)1,2,3,4-tetrahydroisoquinolin-7-amine(3)1,2-diphenylethan-1-amine(15)1,3,4-thiadiazol-2-amine(9)1,3,5-triazin-2-amine(3)1-(2,3-dihydro-1H-inden-5-yl)ethan-1-amine(3)1-(2-aminophenyl)ethan-1-one(18)1-(2-bromophenyl)ethan-1-amine(10)1-(2-chlorophenyl)-1H-pyrazole(3)1-(2-fluorophenyl)ethan-1-amine(40)1-(2-fluorophenyl)propan-1-amine(5)1-(3-(trifluoromethoxy)phenyl)ethan-1-amine(6)1-(3-(trifluoromethyl)phenyl)ethan-1-amine(13)1-(3-aminophenyl)ethan-1-one(11)1-(3-chlorophenyl)-N-methylmethanamine(7)1-(4-(trifluoromethoxy)phenyl)ethan-1-amine(6)1-(anthracen-9-yl)ethan-1-amine(3)1-(pyridin-2-yl)ethan-1-amine(15)1-(pyridin-3-yl)ethan-1-amine(17)1-(pyridin-4-yl)ethan-1-amine(23)1-aminoanthracene-9,10-dione(7)1-benzyl-N-methylpiperidin-3-amine(6)1-benzylpiperidin-4-amine(8)1-methyl-1H-pyrazol-3-amine(14)1-phenyl-1H-pyrazol-3-amine(3)1-phenyl-1H-pyrazol-5-amine(4)1-phenylcyclobutan-1-amine(5)1-phenylcyclopropan-1-amine(26)1-phenylethan-1-amine(257)1H-1,2,4-triazol-3-amine(7)1H-1,2,4-triazol-5-amine(6)1H-benzo[d]imidazol-2-amine(7)1H-benzo[d]imidazol-5-amine(4)1H-benzo[d]imidazol-6-amine(6)1H-imidazol-2-amine(6)1H-indazol-3-amine(23)1H-indazol-4-amine(12)1H-indazol-5-amine(3)1H-indazol-6-amine(5)1H-indazol-7-amine(6)1H-indol-4-amine(9)1H-indol-6-amine(14)1H-indol-7-amine(8)1H-pyrazol-4-amine(6)1H-pyrazol-5-amine(19)1H-pyrrol-1-amine(6)1H-pyrrolo[2,3-b]pyridin-6-amine(4)2,2,2-trifluoro-1-phenylethan-1-amine(38)2,3-dichloroaniline(16)2,3-difluoroaniline(25)2,3-dihydrobenzofuran-3-amine(11)2,3-dihydrobenzofuran-5-amine(3)2,4-dibromoaniline(11)2,4-dichloropyrimidin-5-amine(6)2,4-difluoroaniline(25)2,5-difluoroaniline(23)2,6-dibromoaniline(16)2,6-dichloroaniline(23)2,6-difluoroaniline(15)2,6-diiodoaniline(8)2-((2-nitrophenyl)amino)ethan-1-ol(4)2-((3-phenylpropyl)amino)-1-(pyrrolidin-1-yl)ethan-1-one(2)2-(1H-benzo[d]imidazol-2-yl)ethan-1-amine(5)2-(1H-indol-3-yl)ethan-1-amine(4)2-(diethylamino)ethyl benzoate(3)2-(methylsulfonyl)aniline(3)2-(phenylamino)benzoic acid(11)2-(piperazin-1-yl)ethan-1-amine(6)2-(piperidin-1-yl)ethan-1-amine(8)2-(trifluoromethoxy)aniline(15)2-(trifluoromethyl)aniline(44)2-amino-2-(2-fluorophenyl)ethan-1-ol(3)2-amino-2-phenylethan-1-ol(96)2-amino-3,3-diphenylpropanoic acid(4)2-amino-3-chlorophenol(4)2-amino-3-fluorobenzoic acid(6)2-amino-6-bromophenol(3)2-amino-6-chlorobenzoic acid(5)2-amino-6-chlorobenzonitrile(2)2-amino-6-chlorophenol(4)2-aminobenzamide(17)2-aminobenzenesulfonic acid(8)2-aminophenol(57)2-aminopyrimidin-4(3H)-one(11)2-bromo-3-chloroaniline(4)2-bromo-3-fluoroaniline(6)2-bromo-3-methylaniline(6)2-bromo-4-methylaniline(8)2-bromo-6-methylaniline(9)2-bromo-N,N-dimethylaniline(5)2-bromo-N-methylaniline(8)2-chloro-3-methylaniline(3)2-chloro-4-methylaniline(5)2-chloro-6-methylaniline(10)2-chloro-N-methylaniline(3)2-chloro-N-phenylaniline(8)2-chloroaniline(96)2-chlorobenzene-1,3-diamine(3)2-chlorobenzene-1,4-diamine(4)2-chloropyrimidin-4-amine(22)2-fluoro-6-iodoaniline(7)2-fluoro-N-phenylaniline(4)2-fluoroaniline(113)2-iodo-4-methylaniline(4)2-iodo-N,N-dimethylaniline(3)2-iodoaniline(52)2-methoxypyridin-3-amine(6)2-methyl-6-nitroaniline(6)2-methyl-N-phenylaniline(7)2-methylpyridin-4-amine(7)2-methylpyrimidin-4-amine(23)2-nitro-N-phenylaniline(3)2-phenoxyaniline(4)2-phenylcyclopropan-1-amine(20)2-phenylpyrimidin-5-amine(5)3,4-dibromoaniline(4)3,4-dichloroaniline(13)3,4-difluoroaniline(26)3-(1,3,2-dioxaborinan-2-yl)aniline(3)3-(benzyloxy)aniline(10)3-(methylsulfonyl)aniline(7)3-(piperazin-1-yl)aniline(3)3-(trifluoromethoxy)aniline(12)3-(trifluoromethyl)aniline(65)3-aminobenzenesulfonic acid(4)3-aminodihydrofuran-2(3H)-one(4)3-bromo-2-fluoroaniline(11)3-bromo-4-chloroaniline(8)3-bromo-4-fluoroaniline(15)3-bromo-4-methoxyaniline(6)3-bromo-4-methylaniline(9)3-bromoaniline(90)3-bromobenzene-1,2-diamine(10)3-chloro-2-iodoaniline(3)3-chloro-2-methylaniline(10)3-chloro-4-fluoroaniline(14)3-chloro-4-iodoaniline(3)3-chloro-4-methoxyaniline(5)3-chloro-4-methylaniline(8)3-chlorobenzene-1,2-diamine(4)3-chloropyrazin-2-amine(6)3-fluoro-2-methylaniline(8)3-fluoroaniline(129)3-iodo-2-methylaniline(3)3-iodoaniline(22)3-methoxypyridin-2-amine(5)3-methyl-N-phenylaniline(4)3-methylpyridin-2-amine(7)3-nitroaniline(55)3-phenoxyaniline(2)3-phenyl-1H-pyrazol-5-amine(11)3-phenylisoxazol-5-amine(8)4,6-dichloropyrimidin-2-amine(9)4-(benzyloxy)aniline(5)4-(oxazol-5-yl)aniline(3)4-(pyrrolidin-1-yl)aniline(3)4-(trifluoromethoxy)aniline(12)4-amino-2-chlorobenzonitrile(8)4-amino-2-fluorophenol(7)4-amino-3-chlorobenzoic acid(4)4-amino-3-chlorophenol(4)4-amino-N-phenylbenzenesulfonamide(2)4-aminopyridazin-3(2H)-one(4)4-benzylaniline(7)4-bromo-3-chloroaniline(13)4-bromo-3-fluoroaniline(12)4-bromo-3-methylaniline(5)4-bromo-3-nitroaniline(5)4-bromo-N,N-diphenylaniline(10)4-bromo-N-phenylaniline(6)4-chloro-2-iodoaniline(7)4-chloro-3-fluoroaniline(8)4-chloro-3-iodoaniline(3)4-chloro-3-methylaniline(8)4-chloro-N-phenylaniline(6)4-chloropyrimidin-2-amine(23)4-chloropyrimidin-5-amine(7)4-fluoro-N-phenylaniline(4)4-fluoroaniline(121)4-iodo-N,N-diphenylaniline(3)4-methoxy-N-phenylaniline(5)4-methyl-3-nitroaniline(4)4-methylpyrimidin-2-amine(9)4-methylthiazol-2-amine(7)4-morpholinoaniline(4)4-nitroaniline(21)4-phenoxyaniline(6)4-phenylpyrimidin-2-amine(15)4-phenylthiazol-2-amine(33)5-bromopyrazin-2-amine(13)5-bromothiazol-2-amine(6)5-chloropyrazin-2-amine(9)5-methoxypyridin-3-amine(4)5-methyl-2-nitroaniline(4)5-methylpyridin-2-amine(7)5-methylpyridin-3-amine(4)5-methylthiazol-2-amine(6)5-phenylthiophen-2-amine(5)5H-dibenzo[b,f]azepine(5)6-amino-2H-benzo[b][1,4]oxazin-3(4H)-one(3)6-bromopyrazin-2-amine(5)6-chloropyrazin-2-amine(11)6-chloropyridazin-3-amine(5)6-chloropyrimidin-4-amine(8)6-methoxypyridin-2-amine(14)6-methoxypyridin-3-amine(8)6-methylpyridin-2-amine(46)6-methylpyridin-3-amine(5)6-phenoxypyridin-3-amine(5)7-methoxy-N-phenylquinazolin-4-amine(12)7H-purin-2-amine(6)7H-purin-6-amine(9)7H-pyrrolo[2,3-d]pyrimidin-2-amine(5)7H-pyrrolo[2,3-d]pyrimidin-4-amine(3)9H-purin-6-amine(3)[1,1'-biphenyl]-2-amine(8)[1,1'-biphenyl]-3-amine(12)[1,2,4]triazolo[1,5-a]pyridin-2-amine(17)[1,2,4]triazolo[4,3-a]pyridin-3-ylmethanamine(5)azetidin-3-amine(4)azetidin-3-ylmethanamine(3)aziridine(9)benzo[b]thiophen-2-amine(5)benzo[d]oxazol-2-amine(16)benzo[d]thiazol-2-amine(30)benzo[d]thiazol-5-amine(3)benzo[d]thiazol-6-amine(3)benzyl-3-aminopiperidine-1-carboxylate(6)benzyl-3-aminopyrrolidine-1-carboxylate(7)bis(pyridin-2-ylmethyl)amine(6)chroman-2-ylmethanamine(3)chroman-3-amine(3)chroman-4-amine(20)cyclobutanamine(90)cyclohexanamine(45)cyclopent-2-en-1-amine(4)cyclopentanamine(5)cyclopropyl(phenyl)methanamine(22)diphenylmethanamine(26)ethyl 2-aminobenzoate(13)ethyl 3-aminobenzoate(14)ethyl 4-aminobenzoate(12)imidazo[1,2-a]pyridin-3-amine(7)imidazo[1,2-a]pyridin-8-amine(7)isoquinolin-1-amine(18)isoquinolin-3-amine(13)isoquinolin-4-amine(16)isoquinolin-5-amine(9)isoquinolin-8-amine(6)isoxazol-5-amine(15)methyl 2-amino-2-phenylacetate(26)methyl 2-aminobenzoate(52)methyl 3-amino-3-phenylpropanoate(19)methyl 3-aminobenzoate(49)methyl 4-aminobenzoate(32)methyl L-phenylalaninate(17)morpholin-2-ylmethanamine(8)N,N-diethylaniline(15)N,N-dimethyl-2-phenoxyethan-1-amine(12)N,N-dimethylaniline(63)N,N-dimethylnaphthalen-1-amine(5)N,N-dimethylpyridin-2-amine(10)N,N-dimethylpyridin-4-amine(6)N,N-dimethylpyrimidin-2-amine(10)N,N-dimethylpyrrolidin-3-amine(7)N,N-diphenyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline(6)N,N-diphenyl-[1,1'-biphenyl]-4-amine(8)N-(4-aminophenyl)benzamide(2)N-(furan-2-ylmethyl)aniline(5)N-(prop-2-yn-1-yl)-2,3-dihydro-1H-inden-1-amine(3)N-benzyl-1-phenylethan-1-amine(7)N-benzyl-2-phenylethan-1-amine(1)N-benzylpiperidin-4-amine(3)N-cyclopropylpyrimidin-2-amine(5)N-methyl-1,2-diphenylethan-1-amine(7)N-methyl-1-phenylmethanamine(34)N-methyl-2-nitroaniline(13)N-methyl-4-nitroaniline(3)N-methylaniline(74)N-methylcyclohexanamine(7)N-methylpiperidin-3-amine(5)N-methylpyridin-2-amine(47)N-methylpyridin-3-amine(15)N-methylpyrimidin-2-amine(10)N-methylpyrimidin-4-amine(10)N-methylpyrrolidin-3-amine(6)N-phenyl-3-(trifluoromethyl)aniline(3)N-phenyl-4-(trifluoromethyl)aniline(3)N-phenylnaphthalen-1-amine(5)N-phenylpyridin-2-amine(9)N-phenylpyrimidin-2-amine(5)N-phenylpyrimidin-4-amine(7)N1,N1-diphenylbenzene-1,4-diamine(2)N1-phenylbenzene-1,4-diamine(3)N2,N4,N6-triphenyl-1,3,5-triazine-2,4,6-triamine(4)N2,N4-diphenylpyrimidine-2,4-diamine(5)naphthalen-2-amine(13)O-benzylhydroxylamine(9)oxazol-2-amine(8)phenylmethanamine(154)piperidin-2-ylmethanamine(6)piperidin-3-amine(17)piperidin-3-ylmethanamine(5)piperidin-4-amine(14)piperidin-4-ylmethanamine(7)pyrazin-2-amine(22)pyrazin-2-ylmethanamine(11)pyridazin-3-amine(18)pyridazin-4-amine(10)pyridin-2-amine(29)pyridin-2-ylmethanamine(38)pyridin-3-amine(21)pyridin-3-ylmethanamine(29)pyridin-4-amine(22)pyridin-4-ylmethanamine(12)pyridine-2,3-diamine(5)pyridine-2,6-diamine(12)pyrimidin-2-ylmethanamine(7)pyrimidin-4-amine(29)pyrimidin-5-amine(6)pyrimidine-2,4-diamine(14)pyrrolidin-2-ylmethanamine(12)pyrrolidin-3-ylmethanamine(6)quinazolin-2-amine(13)quinazolin-4-amine(8)quinoxalin-2-amine(3)tetrahydrofuran-3-amine(6)thiazol-2-ylmethanamine(5)thiazol-5-amine(6)thiophen-3-amine(18)tri([1,1'-biphenyl]-4-yl)amine(9)(dimethylamino)but-2-enamide(6)(R)-1-ethanamine(175)(R)-1-propan-1-amine(29)(R)-2,2,2-trifluoro-1-ethanamine(12)(R)-2-amino-acetic acid(26)(R)-2-amino-ethanol(71)(R)-2-amino-propan-1-ol(5)(R)-2-amino-propanoic acid(47)(R)-2-methyl-propan-1-amine(10)(R)-3-amino-propanoic acid(30)(R)-methyl 2-amino-acetate(11)(R)-methyl 2-amino-propanoate(14)(R)-N-methyl-ethanamine(5)(S)-1-ethanamine(191)(S)-2,2,2-trifluoro-1-ethanamine(13)(S)-2-amino-acetic acid(28)(S)-2-amino-ethanol(79)(S)-2-amino-propan-1-ol(7)(S)-2-amino-propanoic acid(117)(S)-2-methoxyethanamine(6)(S)-2-methylpropan-1-amine(8)(S)-3-amino-4-butanoic acid(9)(S)-3-amino-propan-1-ol(2)(S)-butan-1-amine(5)(S)-methyl 2-amino-2-acetate(8)(S)-methyl 2-amino-3-propanoate(22)(S)-methyl 3-amino-propanoate(19)(S)-N-methyl-ethanamine(8)1,2-diamine(9)1-ethanamine(158)1-propan-1-amine(27)2,2,2-trifluoro ethanamine(25)2,2-dimethyl-propan-1-amine(6)2-amino-acetamide(7)2-amino-acetic acid(42)2-amino-acetonitrile(5)2-amino-ethanone(11)2-amino-propan-1-ol(8)2-Amino-propanoic acid(56)2-aminoethanol(34)2-methylpropan-1-amine(8)2-propan-2-amine(31)3-(dimethylamino)prop-2-enoyl(13)3-amino propanoic acid(40)3-amino-1-propanol(6)3-amino-butyric acid(14)3-aminopropanoic acid(40)3-methyl-butan-1-amine(3)3-propan-1-amine(28)amino-2-methylpropan-2-ol(2)amino-alcohol-hydrochloride(78)amino-benzamide(4)amino-benzooxazine(4)amino-benzoxazole(24)amino-carboxylate(969)amino-chromen(50)amino-cyano(5)amino-ethanol(25)amino-imidazole(82)amino-indazole(21)amino-indazole-carboxylate(12)amino-inden(35)amino-methanol(4)amino-methyl-butanol(7)amino-pentane-hydrochloride(144)amino-phenyl-boronic-acid(9)amino-propanol(6)amino-purin(185)amino-pyrazole-carboxylate(66)amino-pyrazolopyrimidine(6)amino-pyrrole-carboxylate(162)amino-quinazoline(93)amino-thiadiazole(24)amino-toluene(47)amino-triazine(31)amino-triazole(58)aminoacetamido(4)aminoacetyl(2)aminoadamantan(5)aminoadamantan(5)aminoazepane(10)aminobenzene(7)aminobenzofuran(5)aminobutanamide(1)aminobutane(23)aminobutane(23)aminobutyl(16)aminobutyl(16)aminocarboxylic acid(21)aminocyclobutanecarboxylate(8)aminocyclohexanecarboxylate(20)aminocyclopentanecarboxylate(10)aminocyclopropanecarboxylate(4)aminoethoxy(18)aminoethoxy(18)aminoethyl(157)aminoethyl(157)aminohexan(6)aminohexyl(3)aminohexyl(3)aminohydroxy(5)aminoindole(6)aminoisonicotinic acid(12)aminoisoquinoline(13)aminomethyl(403)aminomethyl(403)aminomethyl-ol(6)aminooxazole(3)aminooxy(6)aminopentyl(10)aminopentyl(10)aminopiperidine(33)aminopropanamido(1)aminopropane(20)aminopropane(20)aminopropoxy(2)aminopropoxy(2)aminopyrazine(11)aminopyrazolo(5)aminopyridine(62)aminopyrimidine(37)aminopyrrolidine(28)aminoquinoline(47)aminoquinoxalin(1)aminosuccinate(4)aminotetrahydro(12)aminotetrahydro(12)aminotetrahydrofuran(7)aminotetrahydropyran(9)aminothiazole(24)azepin(172)benzimidazol-amine(28)benzopyran-ethylamine(1)benzothiazol-amine(6)benzothiophene-amine(4)benzotriazole-ethylamine(2)benzoxazol-amine(11)benzylaminoethanol(3)benzylazetidine(8)benzylpropan-1-amine(3)benzylpyrazol-amide(4)bromo-indazol-amine(4)bromobenzylamine(6)bromobenzylcarbamate(28)but-3-en-1-amine(6)butan-1-amine(12)butoxycarbonylamino(8)butylamino(7)butylcarbamoyl(5)carbamothioyl(2)carbamoyl(114)carbamoylpiperidine(5)carbazol-amide(6)chloro-indazole-amine(1)chloro-inden-amine(10)chloro-propylamine-hydrochloride(2)chlorobenzylamine(4)chloroethylamino(8)chroman-amine(70)diamino(90)diamino(90)diaminopteridin(3)diaminopteridin(3)diaminopyrimidine(23)dibutylamino(6)dibutylamino(6)dichlorobenzylamine(5)diethanamine(6)diethanamine(6)diethylamino(91)diethylamino(91)diethylaminoethoxy(4)diethylaminoethyl-carboxamide(8)dihydroxy-amino(2)diisopropylamino(1)diisopropylcarbamimidate(2)diisopropylcarbamimidate(2)dimethyl-ethylamine(44)dimethyl-pyrazol-amide(38)dimethylamino(490)dimethylamino(490)dimethylamino ethoxy(25)dimethylamino-propan-1-one(4)dimethylaminomethyl(2)dimethylaminomethyl(2)dimethylaminopropoxy(5)dimethylcarbamoyl(14)dimethylmethanamine(16)dipropylamino(1)dipropylamino(1)enamine-hydrochloride(2)ethanamine(602)ethanamine-hydrochloride(69)ethyl 2-amino-propanoate(5)ethyl 2-aminoacetate(6)ethyl 3-amino-propanoate(5)ethyl aminocarboxylate(5)ethyl-pyrazol-amide(4)ethylamino(37)ethylamino(37)ethylaminoethanol(3)fluindamine(12)fluoren-amine(72)Fmoc(386)hydroxyamino(3)hydroxyamino(3)imidazol-amine(95)iminoacetate(4)indazol-amine(89)inden-amine(104)indene-bromide(5)indol-amine(85)indol-methanamine(17)indole-ethylamine(3)isopropyl-pyrazol-amide(3)isopropylamino(26)isothiazole-amine(6)methanamine(5)methoxy-inden-amine(13)methoxyethanamine(167)methoxyethanamine(167)methoxyimino(2)methyl (R)-3-amino-propanoate(9)methyl 2-amino-propanoate(5)methyl 3-amino-propanoate(9)methyl aminoacetate(4)methyl aminocarboxylate(10)methyl-amine(9)methyl-indanamine(27)methyl-indazolamide(13)methyl-propanoate-hydrochloride(99)methylamine-hydrochloride(4)methylamino(241)methylamino(241)methylbutan-amine(16)methylcarbamoyl(19)methylcarbamoyl(19)methylpropan-2-amine(7)minodiethanol(11)N,N,N-trimethylaminium(4)N,N-dimethyl-ethanamine(10)N,N-dimethyl-methanamine(58)N,N-dimethylpropan-1-amine(6)N-(2-propynyl)-1-amine(6)N-ethyl-amine(37)N-ethyl-N-methylethanamine(9)N-methyl-ethanamine(7)N-methyl-methanamine(105)naphthalen-amine(207)naphthyridin-amine(19)oxadiazol-amide(3)oxy ethanamine(15)pentan-1-amine(5)phenyl-amine(1737)phenyl-amine-hydrochloride(945)phenyl-ethylamine(36)phenyl-methanamine-hydrochloride(113)phenyl-methylamine(238)phenyl-pyrazol-amide(83)phenylamine(572)piperazin-amine(50)piperidin-amine(1)propanediamine(6)propylamino(5)propylamino(5)pyrazin-amine(89)pyrazol-amine(43)pyrazole-methamine(3)pyridazin-amine(55)pyridin-ethanamine(93)pyridin-methanamine(136)pyridinamine(4)pyridine-propylamine(7)pyrimidinamine(2)pyrrol-amine(165)pyrrolopyridine-methylamine(3)quinoline-ethylamine(5)silanamine(3)spiro-amine-hydrochloride(1)tert-butylamino(4)tert-butylaminomethyl(6)tetrahydro-pyran-amine(160)tetrahydrofuran-methylamine(15)tetrahydroisoquinolin-amine(5)tetrahydroquinolin-amine(11)thiadiazol-amine(37)thiazol-amide-hydrochloride(18)thiazol-ethanamine(4)thiophen-ethanamine(8)triamine(25)triamine(25)triazin-amine(60)triazol-amine(94)trifluoromethanamine(1)trifluoromethyl-amine(6)trifluoromethyl-phenyl-ethanamine(29)trifluoromethyl-phenyl-ethylamine(1)trifluoromethyl-phenyl-methanamine(16)trimethyl methanaminium(8)yn-amine-hydrochloride(25)
Aryls(187146)
(6-phenoxytetrahydro-2H-pyran-2-yl)methanol(19)(8S,9S,14R)-1,2,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-3H-cyclopenta[a]phenanthren-3-one(1)(9H-fluoren-9-yl)methyl (2-(tritylthio)ethyl)carbamate(10)(9H-fluoren-9-yl)methyl cyclohexylcarbamate(8)(benzylsulfonyl)benzene(11)(cyclopentyloxy)benzene(12)1,1-dimethylcyclopropane(5)1,2-bis(diphenylphosphaneyl)ethane(4)1,3,4,5-tetrahydro-2H-benzo[b]azepin-2-one(15)1,3-diphenylurea(24)1-(2-hydroxyphenyl)-2-phenylethan-1-one(5)1-(2-hydroxyphenyl)-3-phenylprop-2-en-1-one(10)1-([1,1'-biphenyl]-4-yl)ethan-1-one(5)1-(naphthalen-2-yl)ethan-1-one(3)1-benzyl-4-chlorobenzene(6)1-benzyl-4-fluorobenzene(2)1-bromonaphthalene(4)1-chloronaphthalene(24)1-fluoronaphthalene(23)1-methoxynaphthalene(18)1-methyl-4-(phenylethynyl)benzene(6)1-methylnaphthalene(10)1-naphthaldehyde(23)1-naphthoic acid(23)1-nitronaphthalene(3)10H-phenothiazine(21)2,2',3,3',5,5',6,6'-octafluoro-1,1'-biphenyl(4)2,5-diphenylthiophene(5)2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol(7)2-(trifluoromethyl)-1,1'-biphenyl(8)2-benzylaniline(5)2-bromo-1,1'-biphenyl(12)2-bromonaphthalene(4)2-chloro-1,1'-biphenyl(8)2-chloronaphthalene(6)2-ethoxynaphthalene(4)2-fluorocyclopropane-1-carboxylic acid(7)2-hydroxycyclohexa-2,5-diene-1,4-dione(3)2-iodonaphthalene(5)2-methoxy-1,1'-biphenyl(7)2-methoxynaphthalene(23)2-methoxytetrahydro-2H-pyran(9)2-methyl-1,1'-biphenyl(26)2-methylnaphthalene(13)2-naphthaldehyde(11)2-naphthoic acid(14)2-naphthonitrile(13)2-nitronaphthalene(4)2-phenoxytetrahydro-2H-pyran-4-yl acetate(6)2-phenylnaphthalene(12)3,3'-diphenyl-5,5',6,6',7,7',8,8'-octahydro-1,1'-binaphthalene(13)3,3-diphenyl-3H-benzo[c][1,2]oxathiole 1,1-dioxide(18)3-bromo-1,1'-biphenyl(12)3-chloro-1,1'-biphenyl(12)3-fluoro-1,1'-biphenyl(16)3-methyl-1,1'-biphenyl(12)3-methyl-1,1':4',1''-terphenyl(3)3-phenethylphenol(5)3-phenyl-1,2,4,5-tetrazine(9)3-styrylphenol(11)4-(bromomethyl)-1,1'-biphenyl(12)4-(phenyldiazenyl)benzoic acid(4)4-(phenylsulfonyl)morpholine(13)4-(tert-butyl)-1,1'-biphenyl(5)4-cyclohexylphenol(7)4-iodo-1,1'-biphenyl(9)4-methoxy-1,1'-biphenyl(4)9H-xanthene(7)[1,1'-biphenyl]-2-amine(8)[1,1'-biphenyl]-2-carbonitrile(11)[1,1'-biphenyl]-2-carboxylic acid(20)[1,1'-biphenyl]-2-ol(16)[1,1'-biphenyl]-3-amine(12)[1,1'-biphenyl]-3-carbaldehyde(11)[1,1'-biphenyl]-3-carboxylic acid(31)[1,1'-biphenyl]-3-ol(9)[1,1'-biphenyl]-4-carbaldehyde(27)[1,1'-biphenyl]-4-carbonitrile(22)[1,1'-biphenyl]-4-ol(29)[1,1'-biphenyl]-4-ylboronic acid(14)benzyltriphenylphosphonium(14)cyclopentylbenzene(28)cyclopropanecarboxylic acid(11)cyclopropyl(phenyl)methanamine(22)cyclopropyl(phenyl)methanone(13)diphenyl phosphonate(12)diphenylphosphane(25)diphenylphosphine oxide(25)ethane-1,1-diyldibenzene(3)ethene-1,1,2-triyltribenzene(14)ethyl cyclopropanecarboxylate(8)methyl 1-naphthoate(7)methyl 2-naphthoate(11)methyl [1,1'-biphenyl]-4-carboxylate(8)methyl cyclopropanecarboxylate(8)N,1-diphenylmethanimine(15)N,N-dimethylnaphthalen-1-amine(5)N-((5R,6R)-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-6-yl)-2-phenylacetamide(5)N-benzylbenzamide(21)N-benzylbenzenesulfonamide(17)N-cyclopropylbenzamide(14)N-cyclopropylbenzenesulfonamide(10)N-phenethylbenzamide(9)N-phenyl-2-naphthamide(11)naphthalen-1-ol(28)naphthalen-1-ylboronic acid(4)naphthalen-1-ylmethanol(6)naphthalen-2-amine(13)naphthalen-2-ol(25)naphthalen-2-ylboronic acid(8)naphthalene-1,4-diol(3)naphthalene-2-sulfonic acid(4)propane-2,2-diyldibenzene(17)tert-butyl cyclopropylcarbamate(7)tetrahydro-2H-pyran-2-ol(12)tetrahydro-2H-pyran-2-yl acetate(12)tetraphenylmethane(16)triphenylene(18)vinylcyclopropane(7)acetylbenzonitrile(3)aminobenzyl(7)benzylamine(21)benzylamino(47)benzylhydroxylamine(25)benzylidene(16)benzylidene(16)benzyloxazolidine(1)benzyloxy(1061)benzylpiperazine(14)benzylpiperidine(32)benzylthio(16)benzylthio(16)cyanobenzyl(12)dibenzyl(15)dibenzylamino(7)dibenzylethane(2)dibenzylethane(2)dichlorobenzylidene(8)difluorobenzyl(17)dimethoxybenzyl(20)dimethoxybenzyl(20)dimethylbenzonitrile(25)dimethylbenzyl(6)dimethylbenzyl(6)dimethylbenzylamine(6)ethoxybenzyl(8)ethoxybenzyl(8)fluorobenzyl(132)fluorobenzylidene(2)formylbenzonitrile(35)iodobenzyl(9)isopropylbenzonitrile(4)methoxybenzyl(176)methoxybenzylamine(7)methoxybenzylidene(6)methylbenzonitrile(104)methylbenzyl(74)methylbenzyl(74)methylbenzylamine(8)methylbenzylidene(1)methylbenzylidene(1)nitrobenzyl(33)nitrobenzylidene(2)phenoxybenzonitrile(5)trifluorobenzyl(2)trimethoxybenzyl(7)trimethoxybenzyl(7)
Benzene Compounds(61662)
(((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-fluorophenyl)propanoic acid(3)(10aR)-1,2,3,4,4a,9,10,10a-octahydrophenanthrene(3)(1E,1'E)-N,N'-(ethane-1,2-diyl)bis(1-phenylmethanimine)(5)(1r,4r)-4-phenyl-1,1'-bi(cyclohexane)(24)(9H-fluoren-9-yl)methyl benzylcarbamate(22)(9H-fluoren-9-yl)methyl piperazine-1-carboxylate(7)(cyclopentylmethyl)benzene(5)(R)-3-benzylpyrrolidine(6)(R)-3-phenylmorpholine(7)(R)-4-benzyloxazolidin-2-one(5)(R)-N-(1,2-diphenylethyl)benzenesulfonamide(7)(S)-N-(1,2-diphenylethyl)benzenesulfonamide(7)1,1':2',1''-terphenyl(4)1,1':3',1''-terphenyl(28)1,1':4',1'':4'',1'''-quaterphenyl(7)1,1,2,2-tetra([1,1'-biphenyl]-4-yl)ethene(10)1,1-dimethyl-1,2,3,4-tetrahydronaphthalene(10)1,2,3,4-tetrahydronaphthalen-1-amine(36)1,2,3,4-tetrahydronaphthalen-1-ol(5)1,2,3,4-tetrahydronaphthalene-1-carboxylic acid(3)1,2,3,5,6,7-hexahydro-s-indacene(31)1,2,3,5,6,7-hexahydro-s-indacene(7)1,2,4-trimethylbenzene(22)1,2-dihydroacenaphthylene(6)1,2-dinitrobenzene(4)1,3,5-tris(phenylethynyl)benzene(6)1,3,6,8-tetraphenylpyrene(10)1,3-bis(benzyloxy)benzene(8)1,3-dihydro-2H-inden-2-one(6)1,3-diphenoxypropane(6)1,3-diphenyl-1H-pyrazole(6)1,3-diphenylpropane(7)1,3-diphenylpropane-1,3-dione(4)1,4-bis(phenylethynyl)benzene(7)1,4-diphenylbuta-1,3-diyne(8)1-(2,3-dihydro-1H-inden-5-yl)ethan-1-amine(3)1-(anthracen-9-yl)ethan-1-amine(3)1-benzylazetidine(6)1-bromopyrene(4)1-cyclohexyl-4-(phenylethynyl)benzene(10)1-cyclopropylnaphthalene(6)1-phenyl-1H-tetrazole(6)1-phenylnaphthalene(12)1-trityl-1H-pyrazole(5)1H-indene(19)1H-indene-1,3(2H)-dione(5)2,2 ', 3,3' - tetrahydro-1,1 '- spiro [indene](4)2,2',3,3'-tetrahydro-1,1'-spirobi[indene](9)2,3,4,5-tetrahydro-1H-benzo[c]azepine(7)2,3,5,6,8,9,11,12,14,15-decahydrobenzo[b][1,4,7,10,13,16]hexaoxacyclooctadecine(15)2,3-dihydro-1H-inden-1-amine(52)2,3-dihydro-1H-inden-1-ol(13)2,3-dihydro-1H-inden-4-amine(6)2,3-dihydro-1H-inden-4-ol(6)2,3-dihydro-1H-indene-1-carboxylic acid(5)2,3-dihydro-1H-indene-4-carbonitrile(4)2,4-diphenyl-1,3,5-triazine(6)2,5-diphenyl-1,3,4-oxadiazole(6)2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-fluorene(7)2-(2-chlorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(24)2-(2-fluorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(21)2-(2-methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(29)2-(3-bromophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(14)2-(3-fluorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(19)2-(3-methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(30)2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline(15)2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde(19)2-(4-chlorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(15)2-(4-fluorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(16)2-amino-2-(anthracen-9-yl)ethan-1-ol(4)2-methyl-2,3-dihydro-1H-inden-1-one(9)2-phenyl-1,3,5-triazine(6)2-phenyl-1,3-dioxane(7)2-phenyl-2H-1,2,3-triazole(7)2-phenylisoindoline-1,3-dione(5)2H-benzo[b][1,4]thiazin-3(4H)-one(6)3,3-dimethyl-2,3-dihydro-1H-inden-1-one(5)3,4-dihydro-2H-benzo[b][1,4]dioxepine(7)3,5,6,7-tetrahydro-s-indacen-1(2H)-one(5)3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(2-bromophenyl)propanoic acid(3)3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(2-chlorophenyl)propanoic acid(3)3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(3-(trifluoromethyl)phenyl)propanoic acid(3)3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(3-fluorophenyl)propanoic acid(3)3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(3-methoxyphenyl)propanoic acid(4)3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(3-nitrophenyl)propanoic acid(3)3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-bromophenyl)propanoic acid(3)3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-fluorophenyl)propanoic acid(5)3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-nitrophenyl)propanoic acid(3)3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(o-tolyl)propanoic acid(3)3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(p-tolyl)propanoic acid(3)3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline(22)3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde(19)3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid(12)3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol(13)3-methyl-2,3-dihydro-1H-inden-1-one(4)3-phenyl-1H-pyrrole(5)3-phenylazetidine(5)3-phenyloxazolidin-2-one(4)3-phenylpiperidine(6)4',5'-diphenyl-1,1':2',1''-terphenyl(7)4,4,5,5-tetramethyl-2-(3-(trifluoromethyl)phenyl)-1,3,2-dioxaborolane(19)4,4,5,5-tetramethyl-2-(3-nitrophenyl)-1,3,2-dioxaborolane(24)4,4,5,5-tetramethyl-2-(m-tolyl)-1,3,2-dioxaborolane(21)4,4,5,5-tetramethyl-2-(o-tolyl)-1,3,2-dioxaborolane(24)4,5-diphenyloxazole(7)4,7-diphenylbenzo[c][1,2,5]thiadiazole(6)4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazole(7)4-(phenylethynyl)-1,1'-biphenyl(5)4-bromo-2,3-dihydro-1H-inden-1-one(5)4-bromo-2,3-dihydro-1H-indene(10)4-chloro-2,3-dihydro-1H-inden-1-one(8)4-chloro-2,3-dihydro-1H-indene(5)4-cyclohexyl-1,1'-biphenyl(18)4-fluoro-2,3-dihydro-1H-inden-1-one(6)4-fluoro-2,3-dihydro-1H-indene(14)4-methoxy-2,3-dihydro-1H-inden-1-one(6)4-methyl-2,3-dihydro-1H-inden-1-one(8)4-methyl-2,3-dihydro-1H-indene(3)4-phenyl-1,1'-bi(cyclohexane)(8)4-phenyl-4H-1,2,4-triazole(6)5''-([1,1'-biphenyl]-4-yl)-1,1':4',1'':3'',1''':4''',1''''-quinquephenyl(5)5',5''-diphenyl-1,1':3',1'':3'',1'''-quaterphenyl(8)5,5',6,6',7,7',8,8'-octahydro-1,1'-binaphthalene(6)5,6,7,8-tetrahydronaphthalen-2-ol(5)5-bromo-1,2,3,4-tetrahydronaphthalene(6)5-bromo-2,3-dihydro-1H-indene(8)5-fluoro-2,3-dihydro-1H-indene(3)5-methoxy-1,2,3,4-tetrahydronaphthalene(3)5-methoxy-2,3-dihydro-1H-indene(2)5-methyl-3,4-dihydronaphthalen-1(2H)-one(6)5-phenyl-1H-imidazole(6)5-phenylcyclohexane-1,3-dione(6)6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one(6)6-bromo-1,2,3,4-tetrahydronaphthalene(9)6-bromo-2,3-dihydro-1H-inden-1-one(8)6-bromo-3,4-dihydronaphthalen-1(2H)-one(3)6-chloro-1,2,3,4-tetrahydronaphthalene(3)6-chloro-2,3-dihydro-1H-inden-1-one(5)6-fluoro-1,2,3,4-tetrahydronaphthalene(6)6-fluoro-2,3-dihydro-1H-inden-1-one(4)6-methoxy-2,3-dihydro-1H-inden-1-one(6)6-methoxy-3,4-dihydronaphthalen-1(2H)-one(5)6-methyl-1,2,3,4-tetrahydronaphthalene(5)6-methyl-2,3-dihydro-1H-inden-1-one(3)6-methyl-3,4-dihydronaphthalen-1(2H)-one(2)7-bromo-2,3-dihydro-1H-inden-1-one(13)7-chloro-2,3-dihydro-1H-inden-1-one(9)7-fluoro-2,3-dihydro-1H-inden-1-one(8)7-hydroxy-2,3-dihydro-1H-inden-1-one(6)7-methoxy-2,3-dihydro-1H-inden-1-one(6)7-methoxy-3,4-dihydronaphthalen-1(2H)-one(6)7-methyl-2,3-dihydro-1H-inden-1-one(7)9,10-bis(phenylethynyl)anthracene(5)9,10-diphenylanthracene(16)9-methylanthracene(4)9-phenylxanthylium(7)9H-fluoren-9-one(17)[1,1'-binaphthalen]-2-amine(3)[1,1':4',1''-terphenyl]-4,4''-dicarbaldehyde(7)benzenesulfinic acid(6)benzil(15)Benzo-18-crown-6(4)benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetraone(10)benzyl ((benzyloxy)carbonyl)glycinate(8)benzyl (4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)carbamate(5)benzyl (S)-pyrrolidin-3-ylcarbamate(7)benzyl 3-phenylpropanoate(8)benzyl piperidin-4-ylcarbamate(7)bicyclo[4.2.0]octa-1,3,5-triene(13)cumene(98)dibenzyl phosphonate(5)diphenylmethanimine(10)ethane-1,1,2-triyltribenzene(5)ethylbenzene(84)mesitylene(40)methyl 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate(14)methyl 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate(11)N,2-diphenylacetamide(7)N,N,N-trimethyl-1-phenylmethanaminium(6)N,N-diphenyl-[1,1'-biphenyl]-4-amine(8)N-benzyl-2-phenylethan-1-amine(1)N-benzylcyclopropanamine(6)N-phenylcinnamamide(15)N1,N1,N4,N4-tetraphenylbenzene-1,4-diamine(6)N4,N4,N4',N4'-tetraphenyl-[1,1'-biphenyl]-4,4'-diamine(7)phenanthrene(23)phenanthrene-9,10-dione(8)phenethyldiphenylphosphane(6)phenothiazin-5-ium(8)phenyl (1r,4r)-[1,1'-bi(cyclohexane)]-4-carboxylate(6)phenyl 4-cyclohexylbenzoate(8)tert-butyl (2,3-dihydro-1H-inden-1-yl)carbamate(3)tert-butylbenzene(140)triphenyl phosphate(6)acetamidophenoxy(5)acetamidophenyl(5)acetylbenzoate(7)acetylphenyl(13)acetylphenyl(13)acetylphenylboronic(3)aminobenzoyl(4)aminobiphenyl(4)aminonaphthalene(14)aminophenoxy(19)aminophenyl(239)anisole(4)anthracene(48)Anthracenes(71)benzaldehyde(872)benzamidophenyl(1)benzenedimethanol(4)benzenethiol(8)benzhydrylazetidine(10)benzhydrylazetidine(10)benzhydryloxy(4)benzhydryloxy(4)benziodoxol(2)benzodioxole(5)benzoquinone(6)benzoquinone(6)benzoylbenzoate(4)benzoylphenyl(4)binaphthalene(123)binaphthalene(123)biphenyl(1394)biphenylboronic(4)biphenylboronic(4)boronophenyl(1)boronophenyl(1)bromobenzo(104)bromobenzoyl(6)bromodibenzo(9)bromophenethyl(6)bromophenoxy(34)bromophenyl(989)butoxyphenyl(8)butylaniline(7)butylbenzene(16)butyldiphenylsilyl(7)butylphenol(20)butylphenyl(32)butylphenyl(32)carbamoylphenoxy(12)carbamoylphenyl(8)carboxyphenyl(17)carboxyphenyl(17)chlorobenzenesulfonyl(23)chlorobenzoyl(9)chlorodibenzo(19)chloronaphthalene(36)chlorophenethyl(7)chlorophenoxy(44)chlorophenylacetate(8)chlorophenylacetate acid(1)chlorophenylboronic(5)chlorotrityl(1)cyclopropylaniline(6)cyclopropylbenzoate(5)cyclopropylnaphthalen(10)cyclopropylnaphthalen(10)cyclopropylphenyl(12)cyclopropylphenyl(12)diaminobenzene(7)dibenzaldehyde(19)dibenzene(7)dibenzimidamide(5)dibenzo(108)dibenzo(108)dibenzoate(9)dibenzonitrile(11)dibromobenzene(45)dibromobenzo(14)dibromodibenzo(8)dibromonaphthalene(15)dibromophenanthrene(3)dibromophenoxy(2)dibromophenylboronic(3)dichlorobenzene(38)dichlorobenzenesulfonyl(2)dichlorobenzoyl(4)dichlorophenethyl(5)dichlorophenoxy(16)dichlorophenyl(345)dichlorophenylboronic(5)didehydrodibenz(2)didehydrodibenz(2)diethoxybenzaldehyde(3)diethoxyphenyl(10)diethylaniline(6)diethylbenzamide(7)diethylbenzene(6)diethylbenzene(6)difluorobenzene(57)difluorobenzo(12)difluorobenzoyl(9)difluoromethyl-benzene(6)difluorophenoxy(9)difluorophenyl(403)difluorophenylboronic(11)dihydroanthracene(1)dihydroanthracene(1)dihydrobenzo(189)dihydrobenzo(189)dihydrodibenzo(7)dihydronaphthalene(104)dihydronaphthalene(104)diiodobenzene(12)diiodophenoxy(6)diisopropylaniline(5)diisopropylbenzamide(8)diisopropylbenzene(3)diisopropylbenzene(3)diisopropylphenyl(40)diisopropylphenyl(40)dimesityl(12)dimesityl(12)dimethoxyacetophenone(5)dimethoxyaniline(20)dimethoxybenzaldehyde(40)dimethoxybenzamide(6)dimethoxybenzene(47)dimethoxybenzo(4)dimethoxybenzo(4)dimethoxybenzoate(20)dimethoxybenzohydrazide(3)dimethoxybenzonitrile(11)dimethoxybenzoyl(4)dimethoxybiphenyl(3)dimethoxybiphenyl(3)dimethoxynaphthalene(10)dimethoxynaphthalene(10)dimethoxyphenol(22)dimethoxyphenyl(257)dimethoxyphenylboronic(17)dimethoxyphenylboronic(17)dimethoxystyryl(4)dimethoxytoluene(3)dimethoxytoluene(3)dimethoxytrityl(3)dimethylaminophenyl(4)dimethylanthracene(4)dimethylbenzaldehyde(27)dimethylbenzamide(37)dimethylbenzene(334)dimethylbenzene(334)dimethylbenzenesulfonamide(16)dimethylbenzo(21)dimethylbenzo(21)dimethylbenzoate(27)dimethylbenzoyl(3)dimethylbenzoyl(3)dimethylbromobenzene(2)dimethylnaphthalene(6)dimethylphenol(36)dimethylphenoxy(13)dimethylphenoxy(13)dimethylphenylboronic(6)dimethylphenylboronic(6)dimethylphenylhydrazine(3)dimethylphenylisocyanate(1)dimethylphenylisothiocyanate(4)dimethyltriphenylamine(4)dinaphtho(5)dinitrobenzene(20)diphenol(32)diphenylacetate(6)diphenylamino(27)diphenylaniline(9)diphenylethanamine(4)diphenylethane(19)diphenylethanol(15)diphenylethanone(5)diphenylethene(6)diphenylethyl(7)diphenylethyl(7)diphenylmethyl(8)diphenylmethyl(8)diphenylmethylene(6)diphenylphosphine(204)diphenylphosphine(204)diphenylphosphinobenzamide(9)diphenylphosphoryl(6)diphenylpropane(22)diphenylpyrazine(5)diphenylsilane(2)diphenylurea(4)diphenylurea(4)diylidenebis(3)diylidenebis(3)ethoxyaniline(10)ethoxybenzaldehyde(9)ethoxybenzoate(12)ethoxybenzonitrile(6)ethoxyphenoxy(7)ethoxyphenoxy(7)ethoxyphenyl(46)ethoxyphenyl(46)ethoxyphenylboronic(7)ethylaniline(8)ethylaniline(8)ethylbenzenesulfonamide(338)ethylbenzo(7)ethylbenzo(7)ethylbenzoate(9)ethylphenol(12)ethylphenyl(39)ethylphenyl(39)ethynylaniline(7)ethynylbenzaldehyde(17)ethynylphenyl(35)fluorobenzeneboronic(24)fluoronaphthalene(34)fluorophenoxy(46)formylbenzoate(39)formylphenoxy(4)formylphenoxy(4)formylphenyl(43)formylphenyl(43)formylphenylboronic(9)formylphenylboronic(9)hexahydrobenzo(3)hexahydrobenzo(3)hexylphenyl(3)hexylphenyl(3)homophenylalanine(18)hydroxybenzenesulfonamide(1)hydroxybenzenesulfonate(5)hydroxybenzimidamide(11)hydroxyisophthalaldehyde(6)hydroxyphenethyl(2)hydroxyphenethyl(2)hydroxyphenyl(461)hydroxyphenyl(461)iodobenzo(15)iodobenzoic(54)iodobenzoyl(7)iodobiphenyl(24)iododibenzo(7)iodonaphthalene(9)iodophenoxy(8)isobutoxyphenyl(15)isobutoxyphenyl(15)isobutylbenzene(5)isobutylbenzene(5)isobutylphenyl(9)isophthalamide(12)isophthalate(8)isophthalonitrile(4)isopropoxyaniline(10)isopropoxyaniline(10)isopropoxybenzaldehyde(7)isopropoxybenzoate(7)isopropoxyphenyl(41)isopropoxyphenyl(41)isopropylaniline(18)isopropylbenzaldehyde(6)isopropylbenzamide(9)isopropylbenzenesulfonamide(19)isopropylbenzoate(8)isopropylphenoxy(1035)isopropylphenoxy(1035)isopropylphenyl(58)mercaptobenzo(21)methanobenzo(15)methanobenzo(15)methoxyacetophenone(12)methoxyaniline(74)methoxybenzaldehyde(104)methoxybenzene(1749)methoxybenzene(1749)methoxybenzenesulfonamide(6)methoxybenzenesulfonic(3)methoxybenzenesulfonyl(13)methoxybenzenesulfonyl(13)methoxybenzimidamide(5)methoxybenzo(52)methoxybenzo(52)methoxybenzoate(116)methoxybenzonitrile(67)methoxybenzoyl(12)methoxybenzoyl(12)methoxycarbonylphenylboronic(5)methoxynaphthalene(56)methoxynaphthalene(56)methoxyphenethyl(15)methoxyphenethyl(15)methoxyphenol(72)methoxyphenoxy(41)methoxyphenoxy(41)methoxyphenylacetate(4)methoxyphenylboronic(24)methoxyphenylboronic(24)methoxyphenylhydrazine(4)methoxystyryl(13)methylacetophenone(6)methylaniline(183)methylanisole(3)methylanthracene(5)methylbenzaldehyde(101)methylbenzamide(90)methylbenzamido(3)methylbenzenesulfinamide(4)methylbenzenesulfonamide(81)methylbenzenesulfonic(9)methylbenzenesulfonyl(4)methylbenzenesulfonyl(4)methylbenzimidamide(6)methylbenzoate(193)methylbenzohydrazide(7)methylbenzothioamide(6)methylbenzothioamide(6)methylbenzotrifluoride(5)methylbenzoyl(24)methylbenzoyl(24)methyldibenzo(76)methyldibenzo(76)methyldiphenylamine(3)methyldiphenylphosphine(20)methyldiphenylphosphine(20)methylnaphthalene(34)methylnaphthalene(34)methylphenol(106)methylphenoxy(18)methylphenoxy(18)methylphenyl(529)methylphenylacetonitrile(45)methylphenylboronic(24)methylphenylhydrazine(10)methylpropiophenone(5)methylpropiophenone(5)naphthaldehyde(28)naphthalene(465)naphthalene(465)naphthaleneboronic(15)naphthalenesulfonic(7)naphthamide(15)naphthonitrile(18)naphthoyl(3)naphthoyl(3)nitrobenzenesulfonyl(8)nitrobenzo(54)nitrobenzoyl(13)nitronaphthalene(24)nitrophenethyl(3)nitrophenoxy(43)nitrophenyl(727)nitrophenylboronic(14)nitrotoluene(27)octylbenzene(11)octylbenzene(11)paracyclophane(2)paracyclophane(2)pentafluorobenzene(13)pentylbenzene(5)pentylbenzene(5)pentylbenzoate(2)pentylphenol(4)pentylphenyl(5)pentylphenyl(5)perfluorophenoxy(5)perfluorophenyl(31)phenoxyacetate(5)phenoxyaniline(8)phenoxybenzaldehyde(5)phenoxybenzoate(5)phenoxyethyl(6)phenoxyethyl(6)phenoxymethyl(7)phenoxymethyl(7)phenoxyphenyl(61)phenoxyphenyl(61)phenoxypropan(4)phenylacetamide(34)phenylacetohydrazide(16)phenylacetonitrile(8)phenylacetophenone(5)phenylacetyl(2)phenylacetyl(2)phenylacrylamide(3)phenylalaninol(4)phenylamino(31)phenylaniline(19)phenylanthracene(16)phenylazetidin(3)phenylazetidin(3)phenylbenzamide(23)phenylbenzenesulfonamide(3)phenylbutanoate(12)phenylbutoxy(2)phenylbutoxy(2)phenylcarbamoyl(5)phenylcarbonothioyl(6)phenylcarbonothioyl(6)phenylcyclohexanone(6)phenylcyclopropanecarboxylic(6)phenylcyclopropyl(7)phenylcyclopropyl(7)phenyldiazenyl(4)phenylene(169)phenylene(169)phenylenediamine(13)phenylenedimethanamine(5)phenylethanamine(12)phenylethane(14)phenylethane(14)phenylethanol(10)phenylethanone(20)phenylethoxy(2)phenylethyl(70)phenylethyl(70)phenylethylisocyanate(2)phenylhex(3)phenylimino(1)phenylisocyanate(9)phenylisoserine(4)phenylmethanaminium(1)phenylmethanesulfonyl(2)phenylmethanesulfonyl(2)phenylmethanone(274)phenylmethoxy(4)phenylmethoxy(4)phenylmethyl(8)phenylmethyl(8)phenylnaphthalene(6)phenylnicotinamide(4)phenylpentan(11)phenylpentan(11)phenylpentanamide(16)phenylpropanamide(15)phenylpropyl(20)phenylpyrazolo(8)phenylsulfinyl(3)phenylsulfinyl(3)phenylsulfonyl(89)phenylsulfonyl(89)phenylthieno(68)phenylthio(24)phenylthio(24)phenylthiophene(23)phenylthiophene(23)phenylthiourea(6)phenylurea(3)phenylurea(3)phenylvinyl(9)phthalate(23)phthalimide(12)phthalonitrile(9)propoxybenzaldehyde(4)propoxyphenyl(29)propoxyphenyl(29)propoxyphenylboronic(80)propylbenzoate(2)propylphenyl(26)propylphenyl(26)Pyrenes(20)quaternaphthalene(6)quaterphenyl(15)quaterphenyl(15)styryl(12)sulfamoylphenyl(3)sulfophenyl(4)sulfophenyl(4)terephthalaldehyde(11)terephthalate(13)terphenyl(239)terphenyl(239)tetraaniline(6)tetrabenzaldehyde(7)tetrafluorobenzene(17)tetrahydrobenzo(70)tetrahydrobenzo(70)tetrahydronaphthalene(149)tetrahydronaphthalene(149)tetralone(12)tetramethylaniline(71)tetramethylaniline(71)tetramethylbenzene(9)tetramethylbenzene(9)tetramethylbiphenyl(57)tetramethylbiphenyl(57)tetraphene(3)tetraphene(3)tetraphenyl(11)tetraphenyl(11)toluenesulfonyl(68)toluoyl(2)tolyl(418)tolyl(418)tolylamino(3)tolylazanediyl(4)tolylazanediyl(4)tolylethynyl(3)tolyloxy(19)tolyloxy(19)tolylphosphine(8)tolylphosphine(8)tolylthio(3)tolylthio(3)trianiline(7)tribenzaldehyde(5)tribenzo(27)tribenzo(27)tribromobenzene(7)trichlorobenzene(20)trichlorophenoxy(3)trichlorophenylboronic(6)triethylbenzene(5)trifluorophenyl(56)trifluorophenylboronic(156)triiodobenzene(5)triisopropylbenzene(9)triisopropylphenyl(6)triisopropylphenyl(6)trimethoxybenzaldehyde(3)trimethoxybenzene(43)trimethoxybenzoate(3)trimethoxystyryl(5)trimethylaniline(18)trimethylbenzene(28)trimethylbenzenesulfonamide(4)trimethylbenzoyl(2)trimethylbenzoyl(2)trimethylphenol(6)trimethylphenyl(20)trimethylphenyl(20)trimethylpropiophenone(4)triphenylphosphoranylidene(9)triphenylphosphoranylidene(9)triphenylvinyl(11)trityl(19)trityl(19)tritylamino(2)tritylaziridine(4)tritylaziridine(4)trityloxy(2)tritylthio(3)tritylthio(3)
Benzyl bromides(1063)
1-(bromomethyl)-2-(trifluoromethyl)benzene(10)1-(bromomethyl)-2-chlorobenzene(28)1-(bromomethyl)-2-fluorobenzene(39)1-(bromomethyl)-3-(trifluoromethyl)benzene(12)1-(bromomethyl)-3-chlorobenzene(18)1-(bromomethyl)-3-fluorobenzene(32)1-(bromomethyl)-3-methoxybenzene(17)1-bromo-2-(bromomethyl)benzene(27)2-(2-(bromomethyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(9)2-(bromomethyl)-1,3-difluorobenzene(11)2-(bromomethyl)benzonitrile(10)methyl 2-(bromomethyl)benzoate(26)
Bromides(52662)
(1-bromoethyl)benzene(9)(2,2-dibromovinyl)benzene(6)(2-(4-bromophenyl)ethene-1,1,2-triyl)tribenzene(11)(2-bromo-6-fluorophenyl)methanol(4)(2-bromoethyl)benzene(29)(2-bromophenyl)(phenyl)methanone(3)(2-bromophenyl)boronic acid(31)(2-bromophenyl)hydrazine(7)(2-bromophenyl)methanamine(11)(2-bromophenyl)methanol(27)(3-bromo-2-fluorophenyl)boronic acid(7)(3-bromo-2-methoxyphenyl)boronic acid(6)(3-bromophenyl)boronic acid(25)(3-bromopropyl)benzene(9)(3-bromopyridin-2-yl)methanol(7)(4-bromophenyl)(phenyl)methanone(9)(5-bromopyridin-2-yl)methanamine(5)(5-bromopyridin-2-yl)methanol(5)(bromoethynyl)benzene(5)(dibromomethyl)benzene(6)1,2,3-tribromobenzene(5)1,2,4-tribromobenzene(8)1,2-dibromo-3-chlorobenzene(3)1,2-dibromo-3-fluorobenzene(7)1,2-dibromo-3-methylbenzene(8)1,2-dibromo-4-fluorobenzene(9)1,2-dibromobenzene(40)1,3-dibromo-2,4-difluorobenzene(5)1,3-dibromo-2,4-dimethylbenzene(3)1,3-dibromo-2-chlorobenzene(5)1,3-dibromo-2-fluorobenzene(11)1,3-dibromo-2-iodobenzene(4)1,3-dibromo-2-methoxybenzene(8)1,3-dibromo-2-methylbenzene(9)1,3-dibromo-2-nitrobenzene(3)1,4-dibromo-2-iodobenzene(3)1-(2-bromophenyl)ethan-1-amine(10)1-(2-bromophenyl)ethan-1-one(23)1-(5-bromopyridin-2-yl)piperazine(6)1-(azetidin-1-yl)-2-bromoethan-1-one(3)1-(benzyloxy)-2-bromobenzene(12)1-(benzyloxy)-3-bromobenzene(12)1-(benzyloxy)-4-bromobenzene(8)1-(bromomethyl)-2-iodobenzene(9)1-(bromomethyl)-2-methoxybenzene(12)1-(bromomethyl)-2-methylbenzene(14)1-(bromomethyl)-2-nitrobenzene(21)1-(bromomethyl)-3-methylbenzene(8)1-benzyl-3-bromobenzene(7)1-benzyl-4-bromo-1H-pyrazole(5)1-bromo-2,3-dichlorobenzene(16)1-bromo-2,3-difluorobenzene(53)1-bromo-2,3-dimethoxybenzene(6)1-bromo-2,3-dimethylbenzene(15)1-bromo-2,4-difluorobenzene(37)1-bromo-2-(bromomethyl)benzene(27)1-bromo-2-(difluoromethyl)benzene(5)1-bromo-2-(methylsulfonyl)benzene(4)1-bromo-2-(tert-butyl)benzene(7)1-bromo-2-(trifluoromethoxy)benzene(19)1-bromo-2-chloro-3-fluorobenzene(14)1-bromo-2-chloro-3-methoxybenzene(6)1-bromo-2-chloro-3-methylbenzene(7)1-bromo-2-chloro-4-nitrobenzene(4)1-bromo-2-cyclopropylbenzene(3)1-bromo-2-ethoxybenzene(16)1-bromo-2-ethylbenzene(11)1-bromo-2-fluoro-3-(trifluoromethyl)benzene(4)1-bromo-2-fluoro-3-methoxybenzene(7)1-bromo-2-fluoro-3-methylbenzene(10)1-bromo-2-fluoro-3-nitrobenzene(10)1-bromo-2-fluoro-4-nitrobenzene(6)1-bromo-2-iodobenzene(57)1-bromo-2-isopropylbenzene(10)1-bromo-2-phenoxybenzene(3)1-bromo-3-(trifluoromethyl)benzene(47)1-bromo-3-chloro-2-fluorobenzene(8)1-bromo-3-chloro-2-methylbenzene(3)1-bromo-3-cyclopropylbenzene(6)1-bromo-3-fluoro-2-iodobenzene(4)1-bromo-3-fluoro-2-methoxybenzene(12)1-bromo-3-fluoro-2-methylbenzene(16)1-bromo-3-fluoro-2-nitrobenzene(10)1-bromo-3-iodobenzene(40)1-bromo-3-methoxybenzene(78)1-bromo-3-methyl-2-nitrobenzene(3)1-bromo-3-nitrobenzene(98)1-bromo-3-phenoxybenzene(8)1-bromo-4-(phenylsulfonyl)benzene(7)1-bromo-4-chloro-2-fluorobenzene(14)1-bromo-4-fluoro-2-methoxybenzene(8)1-bromo-4-fluoro-2-methylbenzene(12)1-bromo-4-iodobenzene(26)1-bromo-4-phenoxybenzene(8)1-bromo-4-styrylbenzene(5)1-bromobicyclo[1.1.1]pentane(3)1-bromoisoquinoline(20)1-bromonaphthalene(4)2,3-dibromopyridine(21)2,4-dibromo-1-chlorobenzene(7)2,4-dibromo-1-fluorobenzene(9)2,4-dibromo-1-methoxybenzene(4)2,4-dibromoaniline(11)2,4-dibromophenol(9)2,4-dibromopyridine(5)2,4-dibromothiazole(5)2,5-dibromopyrazine(4)2,5-dibromopyridine(26)2,5-dibromothiazole(5)2,5-dibromothiophene(18)2,6-dibromo-3-fluoropyridine(3)2,6-dibromoaniline(16)2,6-dibromobenzaldehyde(4)2,6-dibromobenzonitrile(3)2,6-dibromophenol(17)2,6-dibromopyrazine(9)2,6-dibromopyridin-4-amine(3)2,6-dibromopyridine(28)2-(2-bromophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(5)2-(2-bromophenyl)acetic acid(17)2-(2-bromophenyl)acetonitrile(10)2-(4-bromophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(8)2-(bromomethyl)furan(3)2-(bromomethyl)pyridine(27)2-amino-3-bromobenzoic acid(7)2-amino-3-bromobenzonitrile(6)2-amino-3-bromophenol(3)2-amino-6-bromophenol(3)2-bromo-1,1'-biphenyl(12)2-bromo-1,3,4-thiadiazole(11)2-bromo-1,3,4-trifluorobenzene(16)2-bromo-1,3,4-trimethylbenzene(4)2-bromo-1,3-dichlorobenzene(11)2-bromo-1,3-difluorobenzene(26)2-bromo-1,3-dimethoxybenzene(6)2-bromo-1,3-dimethylbenzene(25)2-bromo-1,4-dichlorobenzene(9)2-bromo-1,4-difluorobenzene(36)2-bromo-1,4-dimethoxybenzene(8)2-bromo-1,4-dimethylbenzene(12)2-bromo-1-chloro-3-fluorobenzene(8)2-bromo-1-chloro-4-fluorobenzene(13)2-bromo-1-chloro-4-methylbenzene(6)2-bromo-1-chloro-4-nitrobenzene(7)2-bromo-1-fluoro-3-nitrobenzene(6)2-bromo-1-fluoro-4-(trifluoromethyl)benzene(4)2-bromo-1-fluoro-4-methoxybenzene(12)2-bromo-1-fluoro-4-methylbenzene(11)2-bromo-1-fluoro-4-nitrobenzene(17)2-bromo-1-methoxy-3-methylbenzene(3)2-bromo-1-methoxy-4-methylbenzene(4)2-bromo-1-methyl-4-nitrobenzene(7)2-bromo-1H-benzo[d]imidazole(3)2-bromo-1H-imidazole(13)2-bromo-1H-pyrrole(10)2-bromo-3-(trifluoromethyl)pyridine(4)2-bromo-3-chloroaniline(4)2-bromo-3-chloropyridine(25)2-bromo-3-fluoroaniline(6)2-bromo-3-fluoropyridine(26)2-bromo-3-hydroxybenzaldehyde(4)2-bromo-3-methoxybenzaldehyde(4)2-bromo-3-methoxypyridine(7)2-bromo-3-methylaniline(6)2-bromo-3-methylphenol(4)2-bromo-3-methylpyrazine(5)2-bromo-3-methylpyridine(25)2-bromo-3-nitrobenzoic acid(3)2-bromo-3-nitropyridine(5)2-bromo-4-(trifluoromethyl)aniline(4)2-bromo-4-chloro-1-fluorobenzene(10)2-bromo-4-chloro-1-methoxybenzene(4)2-bromo-4-chloro-1-methylbenzene(5)2-bromo-4-chloro-1-nitrobenzene(4)2-bromo-4-chloroaniline(12)2-bromo-4-chlorophenol(8)2-bromo-4-chloropyridine(7)2-bromo-4-fluoro-1-methoxybenzene(9)2-bromo-4-fluoro-1-methylbenzene(16)2-bromo-4-fluoro-1-nitrobenzene(14)2-bromo-4-fluoroaniline(19)2-bromo-4-fluorobenzaldehyde(5)2-bromo-4-fluorobenzoic acid(5)2-bromo-4-fluorobenzonitrile(4)2-bromo-4-fluorophenol(7)2-bromo-4-methoxy-1-nitrobenzene(6)2-bromo-4-methoxyaniline(5)2-bromo-4-methoxybenzaldehyde(4)2-bromo-4-methoxybenzoic acid(4)2-bromo-4-methylaniline(8)2-bromo-4-methylbenzoic acid(4)2-bromo-4-methylpyridine(6)2-bromo-4-methylthiazole(7)2-bromo-4-nitroaniline(8)2-bromo-5-(trifluoromethyl)pyridine(6)2-bromo-5-chloropyrazine(6)2-bromo-5-chloropyridine(22)2-bromo-5-fluoroaniline(13)2-bromo-5-fluorobenzaldehyde(4)2-bromo-5-fluorobenzoic acid(3)2-bromo-5-fluoropyridine(33)2-bromo-5-methoxybenzaldehyde(4)2-bromo-5-methoxypyridine(11)2-bromo-5-methylpyridine(19)2-bromo-5-methylthiazole(7)2-bromo-5-nitropyridine(11)2-bromo-6-(trifluoromethyl)aniline(5)2-bromo-6-(trifluoromethyl)pyridine(6)2-bromo-6-chloroaniline(10)2-bromo-6-chlorophenol(4)2-bromo-6-chloropyrazine(5)2-bromo-6-chloropyridine(23)2-bromo-6-fluoroaniline(13)2-bromo-6-fluorobenzaldehyde(11)2-bromo-6-fluorobenzoic acid(9)2-bromo-6-fluorobenzonitrile(7)2-bromo-6-fluorophenol(8)2-bromo-6-fluoropyridine(21)2-bromo-6-iodoaniline(4)2-bromo-6-methoxyaniline(4)2-bromo-6-methoxyphenol(5)2-bromo-6-methoxypyridine(15)2-bromo-6-methylaniline(9)2-bromo-6-methylbenzoic acid(5)2-bromo-6-methylphenol(7)2-bromo-6-methylpyrazine(5)2-bromo-6-methylpyridine(17)2-bromo-6-nitroaniline(14)2-bromo-6-nitrophenol(9)2-bromo-6-phenylpyridine(4)2-bromo-9,9-diphenyl-9H-fluorene(5)2-bromo-9H-fluorene(15)2-bromo-[1,2,4]triazolo[1,5-a]pyridine(6)2-bromo-N,N-dimethylaniline(5)2-bromo-N-methylaniline(8)2-bromo-N-phenylacetamide(5)2-bromobenzene-1,3-diol(7)2-bromobenzene-1,4-diol(3)2-bromobenzenesulfonyl chloride(8)2-bromobenzo[d]thiazole(28)2-bromodibenzo[b,d]furan(6)2-bromofuran(21)2-bromoimidazo[1,2-a]pyridine(5)2-bromonaphthalene(4)2-bromonicotinic acid(5)2-bromooxazole(7)2-bromopyrazine(15)2-bromopyridin-3-amine(10)2-bromopyridin-3-ol(7)2-bromopyridine(10)2-bromopyridine 1-oxide(6)2-bromopyrimidine(22)2-bromoquinazoline(4)3,4-dibromoaniline(4)3,4-dibromophenol(3)3,5-dibromopyridine(12)3-(bromomethyl)pyridine(18)3-bromo-1,1'-biphenyl(12)3-bromo-1,5-naphthyridine(8)3-bromo-1-methyl-1H-pyrazole(16)3-bromo-1H-pyrrole(8)3-bromo-1H-pyrrolo[2,3-b]pyridine(7)3-bromo-1H-pyrrolo[3,2-b]pyridine(6)3-bromo-2-chlorobenzaldehyde(4)3-bromo-2-chlorobenzoic acid(6)3-bromo-2-chloropyridin-4-amine(3)3-bromo-2-chloropyridine(20)3-bromo-2-fluoroaniline(11)3-bromo-2-fluorobenzaldehyde(9)3-bromo-2-fluorobenzoic acid(10)3-bromo-2-fluorobenzonitrile(9)3-bromo-2-fluorophenol(5)3-bromo-2-fluoropyridine(21)3-bromo-2-hydroxybenzaldehyde(7)3-bromo-2-hydroxybenzoic acid(4)3-bromo-2-hydroxybenzonitrile(3)3-bromo-2-methoxypyridine(20)3-bromo-2-methylpyridine(27)3-bromo-4-chloroaniline(8)3-bromo-4-chlorobenzoic acid(4)3-bromo-4-chlorophenol(4)3-bromo-4-fluoroaniline(15)3-bromo-4-fluorobenzaldehyde(7)3-bromo-4-fluorobenzamide(3)3-bromo-4-fluorobenzoic acid(5)3-bromo-4-fluorophenol(10)3-bromo-4-hydroxybenzaldehyde(5)3-bromo-4-hydroxybenzoic acid(2)3-bromo-4-methoxyaniline(6)3-bromo-4-methoxybenzaldehyde(7)3-bromo-4-methoxybenzoic acid(7)3-bromo-4-methylaniline(9)3-bromo-4-methylbenzoic acid(6)3-bromo-4-methylpyridine(13)3-bromo-5'-phenyl-1,1':3',1''-terphenyl(4)3-bromo-5-chloropyridine(14)3-bromo-5-fluoropyridine(18)3-bromo-5-methylpyridine(14)3-bromo-5-nitropyridine(10)3-bromoaniline(90)3-bromoazetidine(3)3-bromobenzene-1,2-diamine(10)3-bromobenzene-1,2-diol(8)3-bromobenzo[b]thiophene(5)3-bromobenzoic acid(74)3-bromobenzonitrile(36)3-bromofuran(11)3-bromoimidazo[1,2-a]pyridine(26)3-bromoimidazo[1,2-a]pyrimidine(6)3-bromoisoquinoline(10)3-bromoisoxazole(7)3-bromopicolinic acid(9)3-bromopicolinonitrile(13)3-bromopyrazolo[1,5-a]pyrimidine(11)3-bromopyridazine(25)3-bromopyridin-2-amine(30)3-bromopyridin-2-ol(14)3-bromopyridine 1-oxide(8)3-bromopyrrolidine(6)3-bromothiophene(6)4-(bromomethyl)-1,1'-biphenyl(12)4-(bromomethyl)pyridine(8)4-bromo-1,5-naphthyridine(5)4-bromo-1-methyl-1H-pyrazole(14)4-bromo-1-phenyl-1H-imidazole(9)4-bromo-1-phenyl-1H-pyrazole(11)4-bromo-1H-benzo[d]imidazole(10)4-bromo-1H-indole(25)4-bromo-1H-pyrazolo[3,4-b]pyridine(4)4-bromo-1H-pyrrolo[2,3-b]pyridine(9)4-bromo-2-chloro-1-iodobenzene(4)4-bromo-2-chloropyridine(29)4-bromo-2-fluoro-1-iodobenzene(4)4-bromo-2-fluoropyridine(4)4-bromo-2-iodoaniline(8)4-bromo-2-methoxypyridine(4)4-bromo-3-(trifluoromethyl)aniline(5)4-bromo-3-chloroaniline(13)4-bromo-3-fluoroaniline(12)4-bromo-3-methylaniline(5)4-bromo-3-methylpyridine(5)4-bromo-3-nitroaniline(5)4-bromo-N,N-diphenylaniline(10)4-bromo-N-phenylaniline(6)4-bromo-N-phenylbenzenesulfonamide(19)4-bromobenzo[c][1,2,5]thiadiazole(14)4-bromobenzo[d][1,3]dioxole(6)4-bromobenzo[d]thiazole(9)4-bromobenzofuran(3)4-bromoindoline(4)4-bromoisoindolin-1-one(3)4-bromoisoquinolin-1(2H)-one(3)4-bromoisoquinoline(33)4-bromopyridazine(5)4-bromopyridine(31)4-bromopyrimidine(24)4-bromothiazole(10)5-bromo-1,2,3,4-tetrahydroisoquinoline(4)5-bromo-1-methyl-1H-indole(4)5-bromo-1H-1,2,4-triazole(6)5-bromo-1H-benzo[d][1,2,3]triazole(5)5-bromo-1H-benzo[d]imidazole(9)5-bromo-1H-indazole(14)5-bromo-1H-pyrazole(11)5-bromo-1H-pyrazolo[3,4-b]pyridine(11)5-bromo-1H-pyrazolo[3,4-c]pyridine(5)5-bromo-1H-pyrrolo[2,3-b]pyridine(13)5-bromo-2,3-dihydro-1H-indene(8)5-bromo-2,3-dihydrobenzofuran(6)5-bromo-2-chloro-4-methylpyridine(5)5-bromo-2-chloropyridine(40)5-bromo-2-chloropyrimidine(6)5-bromo-2-fluoropyridine(22)5-bromo-2-methoxypyridine(29)5-bromo-2-methylpyridine(23)5-bromo-2-methylpyrimidine(5)5-bromo-2-phenylpyridine(3)5-bromo-4-methylpyridin-2-amine(5)5-bromo-6-chloropyridin-2-amine(3)5-bromo-7H-pyrrolo[2,3-d]pyrimidine(3)5-bromobenzo[b]thiophene(3)5-bromobenzo[d][1,3]dioxole(12)5-bromobenzo[d]oxazole(3)5-bromoimidazo[1,2-a]pyridine(6)5-bromoindoline(8)5-bromoisoindolin-1-one(3)5-bromoisoquinoline(17)5-bromonicotinaldehyde(7)5-bromonicotinic acid(6)5-bromonicotinonitrile(6)5-bromopicolinic acid(11)5-bromopicolinonitrile(13)5-bromopyrazin-2-ol(5)5-bromopyridin-2-amine(35)5-bromopyridin-2-ol(10)5-bromopyridin-3-amine(10)5-bromopyridin-3-ol(6)5-bromoquinoxaline(6)5-bromothiazole(8)6-bromo-1,2,3,4-tetrahydroisoquinoline(4)6-bromo-1,2,3,4-tetrahydroquinoline(6)6-bromo-1-methyl-1H-indole(3)6-bromo-1H-indazole(23)6-bromo-1H-indole(15)6-bromo-1H-pyrazolo[4,3-b]pyridine(4)6-bromo-1H-pyrrolo[2,3-b]pyridine(6)6-bromo-1H-pyrrolo[3,2-b]pyridine(7)6-bromo-1H-pyrrolo[3,2-c]pyridine(5)6-bromo-3,4-dihydro-2H-benzo[b][1,4]oxazine(5)6-bromo-3,4-dihydroisoquinolin-1(2H)-one(4)6-bromo-3,4-dihydroquinolin-2(1H)-one(4)6-bromo-[1,2,4]triazolo[1,5-a]pyridine(12)6-bromo-[1,2,4]triazolo[4,3-a]pyridine(8)6-bromobenzo[b]thiophene(4)6-bromobenzo[d]oxazole(3)6-bromobenzo[d]thiazole(7)6-bromochroman-4-one(6)6-bromoimidazo[1,2-a]pyrimidine(5)6-bromoindoline-2,3-dione(4)6-bromoisoindolin-1-one(4)6-bromoisoquinoline(9)6-bromonicotinaldehyde(8)6-bromonicotinic acid(9)6-bromonicotinonitrile(7)6-bromopicolinic acid(5)6-bromopyrazolo[1,5-a]pyrimidine(7)6-bromopyridin-2-amine(8)6-bromopyridin-3-amine(12)6-bromopyridin-3-ol(13)6-bromoquinazoline(7)6-bromoquinoxaline(9)7-bromo-1,2,3,4-tetrahydroisoquinoline(4)7-bromo-1H-indazole(18)7-bromo-1H-indole(24)7-bromo-1H-pyrrolo[2,3-c]pyridine(4)7-bromo-2,3-dihydrobenzofuran(6)7-bromo-3,4-dihydro-2H-benzo[b][1,4]oxazine(7)7-bromo-3,4-dihydroisoquinolin-1(2H)-one(5)7-bromo-3,4-dihydronaphthalen-1(2H)-one(8)7-bromobenzo[b]thiophene(5)7-bromobenzo[d]thiazole(8)7-bromobenzofuran(6)7-bromochromane(7)7-bromoindoline(3)7-bromoisoquinoline(6)8-bromo-[1,2,4]triazolo[1,5-a]pyridine(6)8-bromochromane(6)8-bromoimidazo[1,2-a]pyridine(15)8-bromoisoquinoline(17)8-bromoquinazoline(8)9-bromoanthracene(5)bromocyclohexane(6)ethyl 2-bromo-2-phenylacetate(6)methyl 2-(2-bromophenyl)acetate(7)methyl 2-bromo-2-phenylacetate(7)methyl 2-bromo-3-nitrobenzoate(2)methyl 2-bromo-5-fluorobenzoate(4)methyl 2-bromo-6-fluorobenzoate(6)methyl 2-bromo-6-methylbenzoate(6)methyl 2-bromobenzoate(44)methyl 3-(bromomethyl)benzoate(14)methyl 3-bromo-4-chlorobenzoate(5)methyl 3-bromo-4-fluorobenzoate(6)methyl 3-bromo-4-methoxybenzoate(8)methyl 3-bromo-4-methylbenzoate(5)methyl 3-bromobenzoate(60)methyl 3-bromopicolinate(9)methyl 4-amino-3-bromobenzoate(5)methyl 5-amino-2-bromobenzoate(4)methyl 6-bromonicotinate(6)N-(2-benzoylphenyl)-2-bromoacetamide(3)tert-butyl 2-(bromomethyl)morpholine-4-carboxylate(3)tert-butyl 3-bromo-1H-indole-1-carboxylate(9)tert-butyl 3-bromopiperidine-1-carboxylate(4)2,2-dibromovinyl(6)2-bromo-acetamide(11)2-bromoethanone(106)2-bromoethoxy(14)2-bromoethyl(58)3-bromopropyl(12)4-bromobutoxy(2)6-bromohexyl(3)benzothiazolyl-bromide(4)benzothiophene-bromide(33)bromindan-one(5)bromo-benzene-diol(6)bromo-benzimidazole(84)bromo-benzocarbazole(5)bromo-benzooxazin-one(11)bromo-carbazole(95)bromo-carboxylate(41)bromo-chloro-benzene(25)bromo-chromen(36)bromo-fluorene(59)bromo-fluoro-indazole(43)bromo-fluoro-nitro-benzene(23)bromo-fluorobenzene(287)bromo-hydroxy(327)bromo-imidazole(91)bromo-imidazopyridine(85)bromo-indazole(173)bromo-indazole-carboxylate(26)bromo-indene(84)bromo-indole-carboxylate(56)bromo-methoxy-indazole(13)bromo-methyl-imidazole(88)bromo-methyl-indole(3)bromo-methyl-pyrazole(169)bromo-methylbenzene(180)Bromo-naphtho-benzofuran(2)bromo-naphthyridin-one(5)bromo-naphthyridine(61)bromo-nitrobenzene(188)bromo-pyrazol-amide(7)bromo-pyrazole-carboxylate(60)bromo-pyrazolopyridine(117)bromo-pyrrolopyridine(169)bromo-pyrrolopyrimidine(28)bromo-thiadiazole(58)bromo-triazole(143)bromo-trifluoromethyl-pyridine(36)bromoacetate(3)bromoacetyl(14)bromoaniline(2)bromoanthracene(5)bromoanthracene(5)bromobenzaldehyde(15)bromobenzamide(7)bromobenzene(24)bromobenzimidamide(6)bromobenzofuran(17)bromobenzoic(13)bromobenzotrifluoride(2)bromobenzyl(36)bromobut(2)bromobut(2)bromobutoxy(4)bromobutoxy(4)bromobutyl(6)bromobutyl(6)bromochroman(26)bromochroman(26)bromocinnoline(5)bromocyclobutanecarboxylate(6)bromocyclopropanecarboxylate(11)bromoethoxy(22)bromoethoxy(22)bromoethyl(86)bromoethyl(86)bromoethynyl(6)bromohexyl(11)bromohexyl(11)bromoindole(8)bromoisobenzofuran(1)bromomethyl(760)bromomethyl-indazole(23)bromonaphthalene(33)bromonicotinaldehyde(5)bromonicotinamide(5)bromooctyl(3)bromooxazole(6)bromopentyl(8)bromopentyl(8)bromophenyl-boronate(2)bromophthalazin(11)bromopiperidine(6)bromopropane(10)bromopropane(10)bromopropoxy(6)bromopropoxy(6)bromopropyl(25)bromopropyl(25)bromopyrazine(25)bromopyrazolo(34)bromopyridazine(9)bromopyridine(209)bromopyridine-boronate(4)bromopyrimidine(47)bromopyrrole-carboxylate(61)bromoquinazoline(19)bromoquinoline(119)bromoquinoxaline(8)bromothiazole(32)bromothiochroman(4)bromothiochroman(4)bromothiophene(72)bromovinyl(2)dibromo(698)dibromo(698)dibromoanthracene(3)dibromoanthracene(3)dibromobenzaldehyde(8)dibromobenzonitrile(8)dibromomethyl(6)dibromomethyl(6)dibromopropoxy(3)dibromopropoxy(3)dibromoquinoline(6)ethyl 2-bromo-acetate(6)fluoren-one(31)indene-bromide(5)methyl 2-bromo-acetate(7)tert-butoxybromobenzene(6)tert-butylbromobenzene(77)tetrabromo(10)tetrabromo(10)tetrahydroisoquinoline-bromide(6)tetrahydroquinoline-bromide(14)tribromo(9)tribromo(9)tribromomethyl(2)
Carbamates(5359)
(R)-3-(boc-amino)-3-propanoic acid(24)(R)-tert-butyl 2-hydroxyethyl-carbamate(3)(R)-tert-butyl ethylcarbamate(4)(S)-2-((tert-butoxycarbonyl)amino)-acetic acid(12)(S)-2-(boc-amino)(78)(S)-3-(boc-amino)-3-propanoic acid(23)(S)-3-(boc-amino)-4-butanoic acid(24)(S)-methyl 2-((tert-butoxycarbonyl)amino)-propanoate(12)(S)-tert-butyl (3-hydroxypropan-2-yl)carbamate(6)(S)-tert-butyl 2-hydroxyethyl-carbamate(6)(S)-tert-butyl ethylcarbamate(8)(tert-butoxycarbonyl)amino)-acetic acid(12)(tert-butoxycarbonyl)amino)ethyl(8)(tert-butoxycarbonyl)amino-acetic acid(13)2-((tert-Butoxycarbonyl)amino)-propanoic acid(10)2-(boc-amino)-propanoic acid(48)2-(tert-butoxycarbonyl)aminobutanoic acid(5)3-(boc-amino)-3-propanoic acid(25)3-(boc-amino)-butanoic acid(25)bis(tert-butoxycarbonyl)amino(12)boc-amino(85)boc-amino-carboxylic acid(12)boc-aminoethyl(20)Boc-aminomethyl(130)ethyl carbamate(7)ethyl(methyl)carbamate(5)methoxycarbonylamino(15)methyl ((tert-butoxycarbonyl)amino)-carboxylate(6)tert-butyl butylcarbamate(7)tert-butyl hydroxymethylcarbamate(3)tert-butyl methyl(methyl)carbamate(8)tert-butyl methylcarbamate(6)tert-butyl N-methylcarbamate(29)tert-butyl oxyethylcarbamate(6)
Carboxylic Acids(56630)
1,2,3,4-tetrahydroisoquinoline-1-carboxylic acid(19)1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid(12)1,5-naphthyridine-3-carboxylic acid(6)1-((benzyloxy)carbonyl)piperidine-2-carboxylic acid(5)1-((benzyloxy)carbonyl)piperidine-3-carboxylic acid(3)1-((benzyloxy)carbonyl)pyrrolidine-3-carboxylic acid(6)1-methyl-1H-indole-2-carboxylic acid(8)1-naphthoic acid(23)1-phenyl-1H-imidazole-4-carboxylic acid(4)1-phenyl-1H-pyrazole-3-carboxylic acid(9)1-phenyl-1H-pyrazole-4-carboxylic acid(17)1-phenyl-1H-pyrazole-5-carboxylic acid(3)1-phenylcyclobutane-1-carboxylic acid(16)1-phenylcyclohexane-1-carboxylic acid(5)1-phenylcyclopropane-1-carboxylic acid(20)1H-benzo[d]imidazole-2-carboxylic acid(7)1H-benzo[d]imidazole-4-carboxylic acid(6)1H-benzo[d]imidazole-5-carboxylic acid(4)1H-imidazole-2-carboxylic acid(7)1H-imidazole-4-carboxylic acid(4)1H-indazole-3-carboxylic acid(24)1H-indazole-4-carboxylic acid(8)1H-indazole-5-carboxylic acid(4)1H-indazole-7-carboxylic acid(4)1H-indole-3-carboxylic acid(26)1H-indole-4-carboxylic acid(9)1H-indole-5-carboxylic acid(4)1H-indole-6-carboxylic acid(5)1H-indole-7-carboxylic acid(7)1H-pyrazole-4-carboxylic acid(6)1H-pyrazole-5-carboxylic acid(10)1H-pyrrole-2-carboxylic acid(28)1H-pyrrolo[2,3-b]pyridine-3-carboxylic acid(5)1H-pyrrolo[2,3-c]pyridine-2-carboxylic acid(4)1H-pyrrolo[3,2-b]pyridine-3-carboxylic acid(4)2,3-difluorobenzoic acid(16)2,3-dihydrobenzo[b][1,4]dioxine-2-carboxylic acid(3)2,3-dihydrobenzo[b][1,4]dioxine-6-carboxylic acid(3)2,4-difluorobenzoic acid(15)2,5-dichlorobenzoic acid(10)2,5-difluorobenzoic acid(17)2,6-dichlorobenzoic acid(9)2,6-difluorobenzoic acid(25)2-(1,3-dioxoisoindolin-2-yl)propanoic acid(3)2-(1H-indol-1-yl)acetic acid(5)2-(1H-indol-3-yl)acetic acid(23)2-(2-bromophenyl)acetic acid(17)2-(2-chlorophenyl)acetic acid(19)2-(2-fluorophenyl)acetic acid(34)2-(2-hydroxyphenyl)acetic acid(5)2-(2-methoxyphenyl)acetic acid(9)2-(2-methyl-1H-indol-3-yl)acetic acid(5)2-(2-nitrophenyl)acetic acid(15)2-(2-oxo-2H-chromen-4-yl)acetic acid(4)2-(3-fluorophenyl)acetic acid(28)2-(3-hydroxyphenyl)acetic acid(4)2-(3-methoxyphenyl)acetic acid(14)2-(4-chlorophenoxy)acetic acid(2)2-(4-fluorophenyl)acetic acid(17)2-(4-methoxyphenyl)acetic acid(17)2-(4-nitrophenyl)acetic acid(7)2-(benzofuran-3-yl)acetic acid(3)2-(m-tolyl)acetic acid(9)2-(morpholin-2-yl)acetic acid(5)2-(o-tolyl)acetic acid(9)2-(p-tolyl)acetic acid(5)2-(phenylamino)benzoic acid(11)2-(pyridin-3-yl)acetic acid(7)2-(pyrrolidin-1-yl)benzoic acid(3)2-(pyrrolidin-2-yl)acetic acid(8)2-(pyrrolidin-3-yl)acetic acid(6)2-(thiazol-4-yl)acetic acid(7)2-(trifluoromethoxy)benzoic acid(4)2-(trifluoromethyl)benzoic acid(24)2-amino-2-(2-fluorophenyl)acetic acid(11)2-amino-3,3-diphenylpropanoic acid(4)2-amino-3-(2-fluorophenyl)propanoic acid(15)2-amino-3-bromobenzoic acid(7)2-amino-3-chlorobenzoic acid(4)2-amino-6-chlorobenzoic acid(5)2-benzoylbenzoic acid(9)2-bromo-4-fluorobenzoic acid(5)2-bromo-4-methylbenzoic acid(4)2-bromo-6-fluorobenzoic acid(9)2-bromo-6-methylbenzoic acid(5)2-chloro-3-methoxybenzoic acid(4)2-chloro-3-methylbenzoic acid(4)2-chloro-3-nitrobenzoic acid(4)2-chloro-4-fluorobenzoic acid(5)2-chloro-4-methylbenzoic acid(3)2-chloro-6-fluorobenzoic acid(6)2-chlorobenzoic acid(57)2-cyclohexylacetic acid(12)2-fluoro-4-methylbenzoic acid(6)2-fluoro-5-methoxybenzoic acid(5)2-fluorobenzoic acid(71)2-fluorocyclopropane-1-carboxylic acid(7)2-iodobenzoic acid(40)2-methoxyisonicotinic acid(4)2-methyl-2-phenylpropanoic acid(10)2-methyl-3-nitrobenzoic acid(5)2-naphthoic acid(14)2-nitrobenzoic acid(47)2-oxo-2-phenylacetic acid(9)2-oxo-2H-chromene-3-carboxylic acid(14)2-phenoxypropanoic acid(11)3-((tert-butoxycarbonyl)amino)-4-(2-fluorophenyl)butanoic acid(8)3-(1H-indol-3-yl)propanoic acid(4)3-(2-fluorophenyl)acrylic acid(5)3-(3-chlorophenyl)acrylic acid(6)3-(3-chlorophenyl)propanoic acid(4)3-(3-methoxyphenyl)propanoic acid(7)3-(benzyloxy)benzoic acid(5)3-(chlorosulfonyl)benzoic acid(12)3-(methylsulfonyl)benzoic acid(5)3-(phenyldiazenyl)benzoic acid(7)3-(piperazin-1-yl)benzoic acid(3)3-(trifluoromethoxy)benzoic acid(8)3-(trifluoromethyl)benzoic acid(21)3-benzamidopropanoic acid(3)3-bromo-2-chlorobenzoic acid(6)3-bromo-2-fluorobenzoic acid(10)3-bromo-2-hydroxybenzoic acid(4)3-bromo-4-chlorobenzoic acid(4)3-bromo-4-fluorobenzoic acid(5)3-bromo-4-hydroxybenzoic acid(2)3-bromo-4-methoxybenzoic acid(7)3-bromo-4-methylbenzoic acid(6)3-bromobenzoic acid(74)3-chloro-2-fluorobenzoic acid(7)3-chloro-2-methoxybenzoic acid(4)3-chloro-4-fluorobenzoic acid(6)3-chloro-4-methoxybenzoic acid(4)3-chlorobenzoic acid(85)3-fluoro-4-methoxybenzoic acid(5)3-fluorobenzoic acid(103)3-fluoropicolinic acid(6)3-iodobenzoic acid(22)3-methoxy-4-nitrobenzoic acid(3)3-methylpicolinic acid(9)3-nitrobenzoic acid(61)3-phenoxybenzoic acid(6)3-phenoxypropanoic acid(6)4-(1H-pyrazol-1-yl)benzoic acid(3)4-(benzyloxy)benzoic acid(5)4-(phenyldiazenyl)benzoic acid(4)4-(trifluoromethyl)nicotinic acid(3)4-amino-3-chlorobenzoic acid(4)4-benzamidobenzoic acid(3)4-cyclohexylbenzoic acid(6)4-fluoro-3-iodobenzoic acid(5)4-fluorobenzoic acid(56)4-methylnicotinic acid(5)4-morpholinobenzoic acid(5)4-nitrobenzoic acid(17)4-oxo-1,4-dihydroquinoline-3-carboxylic acid(3)4-oxo-4-(phenylamino)butanoic acid(6)4-oxo-4-phenylbutanoic acid(8)4-oxopiperidine-2-carboxylic acid(3)4-phenoxybenzoic acid(4)4-phenylbutanoic acid(12)5'-(4-carboxyphenyl)-[1,1':3',1''-terphenyl]-4,4''-dicarboxylic acid(7)5-oxotetrahydrofuran-2-carboxylic acid(4)5-phenyl-1H-pyrazole-3-carboxylic acid(12)5-phenylfuran-2-carboxylic acid(6)5-phenylisoxazole-3-carboxylic acid(15)6,6-dimethylmorpholine-3-carboxylic acid(5)6-methoxypicolinic acid(7)6-methylnicotinic acid(6)6-methylpicolinic acid(11)6-oxopiperidine-2-carboxylic acid(3)6-phenylpicolinic acid(3)7-fluoro-1H-indole-2-carboxylic acid(5)[1,1'-biphenyl]-2-carboxylic acid(20)[1,1'-biphenyl]-3-carboxylic acid(31)azetidine-2-carboxylic acid(6)benzo[b]thiophene-2-carboxylic acid(32)benzo[d]thiazole-6-carboxylic acid(5)benzofuran-2-carboxylic acid(21)benzofuran-3-carboxylic acid(6)benzoic acid(1529)benzoylglycine(13)bicyclo[1.1.1]pentane-1-carboxylic acid(6)chromane-2-carboxylic acid(6)chromane-3-carboxylic acid(4)cyclobutanecarboxylic acid(23)cyclohex-1-ene-1-carboxylic acid(3)cyclohex-3-ene-1-carboxylic acid(7)cyclohexanecarboxylic acid(54)cyclopentanecarboxylic acid(26)cyclopropanecarboxylic acid(11)ethyl 5-oxopyrrolidine-2-carboxylate(3)furan-2-carboxylic acid(29)furan-3-carboxylic acid(9)imidazo[1,2-a]pyridine-2-carboxylic acid(21)imidazo[1,2-a]pyridine-3-carboxylic acid(14)imidazo[1,2-a]pyridine-5-carboxylic acid(3)imidazo[1,2-a]pyridine-7-carboxylic acid(3)imidazo[1,2-a]pyridine-8-carboxylic acid(6)imidazo[1,2-a]pyrimidine-3-carboxylic acid(5)indoline-2-carboxylic acid(3)isonicotinic acid(23)isoquinoline-1-carboxylic acid(10)isoquinoline-3-carboxylic acid(7)isoquinoline-4-carboxylic acid(12)isoquinoline-5-carboxylic acid(5)isoquinoline-6-carboxylic acid(3)isoxazole-3-carboxylic acid(8)isoxazole-5-carboxylic acid(7)methyl benzofuran-2-carboxylate(6)methyl isoquinoline-3-carboxylate(3)morpholine-2-carboxylic acid(12)morpholine-3-carboxylic acid(10)nicotinic acid(53)oxazole-4-carboxylic acid(4)picolinic acid(22)piperidine-2-carboxylic acid(26)piperidine-3-carboxylic acid(14)piperidine-4-carboxylic acid(12)proline(13)pyrazine-2-carboxylic acid(34)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid(7)pyridazine-3-carboxylic acid(7)pyridine-2,6-dicarboxylic acid(9)pyrimidine-2-carboxylic acid(11)pyrimidine-4-carboxylic acid(17)pyrimidine-5-carboxylic acid(7)pyrrolidine-3-carboxylic acid(14)tetrahydrofuran-2-carboxylic acid(4)tetrahydrofuran-3-carboxylic acid(4)thiazole-2-carboxylic acid(9)thiazole-4-carboxylic acid(5)thiazole-5-carboxylic acid(10)thiophene-3-carboxylic acid(8)tryptophan(23)(R)-2-amino-acetic acid(26)(R)-2-amino-propanoic acid(47)(R)-2-hydroxy-acetic acid(7)(R)-2-hydroxypropanoic acid(2)(R)-3-(boc-amino)-3-propanoic acid(24)(R)-3-amino-propanoic acid(30)(S)-2-((tert-butoxycarbonyl)amino)-acetic acid(12)(S)-2-amino-acetic acid(28)(S)-2-amino-propanoic acid(117)(S)-2-hydroxy-acetic acid(8)(S)-3-(boc-amino)-3-propanoic acid(23)(S)-3-(boc-amino)-4-butanoic acid(24)(S)-3-amino-4-butanoic acid(9)(S)-methyl 2-amino-3-propanoate(22)(tert-butoxycarbonyl)amino)-acetic acid(12)(tert-butoxycarbonyl)amino-acetic acid(13)2,2-difluoroacetic acid(9)2-((tert-Butoxycarbonyl)amino)-propanoic acid(10)2-(boc-amino)-propanoic acid(48)2-(tert-butoxycarbonyl)aminobutanoic acid(5)2-amino-acetic acid(42)2-Amino-propanoic acid(56)2-cyano-acrylic acid(5)2-hydroxyacetic acid(15)2-hydroxypropanoic acid(5)2-methyl-2-oxypropanoic acid(8)2-methylpropanoic acid(27)2-oxopropanoic acid(3)2-oxy-propanoic acid(9)2-propanoic Acid(11)2-propionic acid(38)2-thioacetic acid(13)3-(boc-amino)-3-propanoic acid(25)3-(boc-amino)-butanoic acid(25)3-(oxy)propanoic acid(13)3-amino propanoic acid(40)3-amino-butyric acid(14)3-aminopropanoic acid(40)3-methyl-butanoic acid(5)4-amino-4-oxobutanoic acid(13)4-butanoic acid(39)4-oxo-butanoic acid(25)5-oxo-pentanoic acid(6)acetic acid(643)aceticacid(1249)acetylbenzoic(8)acetylbenzoic acid(3)acrylic(95)acrylic acid(131)amino-2-oxoacetic acid(7)aminobenzoic(7)aminobutanoic(8)aminocarboxylic acid(21)aminoheptanoic(6)aminohexanoic(10)aminoisoquinoline-carboxylic-acid(4)aminonicotinic(4)aminonicotinic acid(3)aminooctanoic(3)aminopentanedioic(18)aminopentanoic(8)aminophenylacetic(21)aminophenylacetic acid(11)aminophthalic(12)aminopropanoic(11)aminoundecanoic(5)aminoundecanoic acid(4)benzenedicarboxylic(6)benzo-carboxylic acid(194)benzoic(682)benzylpropanoic(61)benzylpropanoic acid(61)benzylsuccinic(9)benzylsuccinic acid(3)boc-amino-carboxylic acid(12)boronobenzoic(4)bromobenzoic(13)bromobutanoic(5)bromododecanoate(3)bromoisonicotinic(4)bromoisonicotinic acid(4)bromonicotinic(9)bromonicotinic acid(3)bromooctanoate(1)bromophenylacetic(26)bromophenylacetic acid(7)bromovalerate(4)butanoic(124)butanoic acid(5)carboxylate(10)carboxylic acid-hydrochloride(68)chlorindazole-carboxylic-acid(6)chlorobenzoic(35)chloroisophthalic(1)chloromandelic(6)chloromandelic acid(1)chloropropanoic(1)chroman-carboxylic(26)chromene-carboxylic acid(27)cinnamic acid(5)cyanonicotinic(5)cyanopropanoic(2)cyclobutanecarboxylic(9)cyclopropanecarboxylic(23)diacetic(19)diacetic(19)diaminobenzoic(7)diaminobutanoic(5)diaminohexanoic(4)dibenzoic(40)dibromobenzoic(10)dibromobenzoic acid(4)dibromonicotinic(15)dibromonicotinic acid(5)dicarboxylic(183)dicarboxylic(183)dicarboxylic acid(7)dichlorobenzoic(22)dichlorobenzoic acid(12)dichloroisonicotinic(6)dichloroisonicotinic acid(3)dichlorophenylacetic(21)dichlorophenylacetic acid(2)dienoate(14)dienoic(10)difluorobenzoic(54)difluorobutanoic(3)difluoroisonicotinic(7)difluoroisonicotinic acid(4)difluorophenylacetic(7)difluorophenylacetic acid(5)difluorophthalic(6)difluoropropanoic(51)diiodobenzoic(5)diisophthalic(20)diisophthalic acid(1)diisopropylbenzoic(9)diisopropylbenzoic acid(2)dimethoxybenzoic(33)dimethoxyphenylacetic(6)dimethoxyphenylacetic acid(2)dimethyl-pyrazole-carboxylic acid(13)dimethylbenzoic(32)dimethylbutanoic(13)dimethylbutanoic(13)dimethylpropanoic(5)dinitrobenzoic(3)diphenylbutanoic(2)diphenylphosphinic(2)diphenylpropanoic(8)dipropionate(7)diyldimethanol(17)dodecanoic(25)dodecanoic(25)enedioic(9)enoic(127)ethoxybenzoic(9)ethyl 2,2-difluoro-acetate(8)ethylbenzoic(14)ethylbenzoic acid(4)ethynylbenzoic(6)ethynylbenzoic acid(1)fluorene-carboxylic-acid(119)fluorindole-carboxylic-acid(4)fluoro-indazole-carboxylic-acid(3)fluorobenzoic(82)fluorocarboxylic acid(6)fluoroisophthalic(7)fluorophenylacetic(10)fluorophenylacetic acid(3)fluorophthalic(5)fluorophthalic acid(1)fluoropicolinic(12)formylbenzoic(23)formylbenzoic acid(7)guanidinopentanoic(8)heptanoic(6)heptanoic(6)hexacarboxylic(8)hexacarboxylic(8)hexacarboxylic acid(2)hexanoic(25)hexanoic(25)hexanoic acid(7)hydrazinylbenzoic(6)hydrazinylbenzoic acid(2)hydroxy-carboxylic acid(9)hydroxyacetate(7)hydroxycyclobutanecarboxylate(4)hydroxycyclohexanecarboxylate(10)hydroxynicotinic(15)imidazole-carboxylic acid(53)indazole-carboxylic acid(66)indene-carboxylic acid(14)indolecarboxylic(206)isonicotinic(8)isophthalic(31)isophthalic acid(6)isopropoxybenzoic(9)isopropoxybenzoic acid(3)isopropylbenzoic(13)isopropylnicotinic(7)isopropylnicotinic acid(2)isoquinolinecarboxylic(2)isothiazole-carboxylic-acid(4)isoxazolecarboxylic(48)malonate(39)mercaptobenzoic(6)mercaptobenzoic acid(4)mercaptopropanoic(11)methoxy-acetic acid(5)methoxybenzoic(142)methoxybutanoic(4)methoxybutanoic(4)methoxyindazole-carboxylic-acid(2)methoxyisophthalic(10)methoxyisophthalic acid(2)methoxyphenylacetic(11)methoxyphenylacetic acid(8)methoxypropanoic(7)methoxypropanoic(7)methyl acetate(79)methyl-carboxylic acid(33)methylbenzoic(175)methylbutanoic(32)methylbutanoic(32)methylcinnamic(8)methylcinnamic acid(3)methylhexanoic(8)methylhexanoic(8)methylisophthalic(19)methylisophthalic acid(4)methylnicotinic(46)methyloctanoic(6)methylpentanoic(108)methylpentanoic(108)methylpropanoic(57)methylsuccinic(10)naphthoic(64)naphthyridine-carboxylic acid(33)nitrobenzoic(161)nitroisonicotinic(7)nitroisonicotinic acid(1)nitroisophthalic(7)nitroisophthalic acid(1)nitronicotinic(10)nitrophthalic(3)nitropicolinic(11)octanoic(6)octanoic(6)oxadiazole-carboxylic acid(2)oxoacetic(21)oxobutanoic(57)oxobutanoic(57)oxodecanoic(5)oxodecanoic(5)oxododecanoic(9)oxododecanoic(9)oxoheptanoic(6)oxoheptanoic(6)oxoheptanoic acid(4)oxohexadecanoic(13)oxohexadecanoic(13)oxohexanoic(10)oxohexanoic(10)oxooctadecanoic(7)oxooctadecanoic(7)oxopentanoic(37)oxopentanoic(37)oxopropanoic(8)oxyacetic acid(81)pentanedioic(18)pentanedioic(18)pentanoic(27)pentanoic acid(10)phenoxyacetic(2)phenoxybenzoic(5)phenoxypropanoic(30)phenylacetic(35)phenylbutanoic(47)phenylpentanoic(11)phenylpropanoic(45)picolinic(30)piperidine-carboxylic acid(170)piperidinecarboxylate(8)propanoic(1168)propanoic acid(167)propenoic(6)propiolic(6)propiolic acid(11)propylbenzoic(49)pyrazinecarboxylic(69)pyrazole-carboxylic acid(280)pyridazinecarboxylic(36)pyridinecarboxylic(18)pyrrole-carboxylic acid(40)pyrrolidine-carboxylic acid(269)quinazoline-carboxylic-acid(4)quinoline-carboxylic acid(381)quinoxaline-carboxylic-acid(6)succinic(13)sulfamoylbenzoic(15)sulfobenzoic(5)sulfopropanoic(1)sulfopropanoic(1)terephthalic(7)tetraacetic(5)tetrabenzoic(16)tetrabenzoic acid(3)tetracarboxylic(13)tetrahydrofuran-carboxylic-acid(2)tetrahydropyran-carboxylic-acid(4)thiazole-carboxylic acid(114)thiazolidine-carboxylic(15)thiomorpholine-carboxylic-acid(2)thiophenecarboxylic(153)thiopropanoic acid(4)triazole-carboxylic(5)triazole-carboxylic-acid(1)tribenzoic(18)tribenzoic acid(5)tricarboxylic(18)trichlorobenzoic(5)trichlorobenzoic acid(2)trifluoroacetic(2)trifluorobenzoic(12)trifluorobenzoic acid(5)trifluorobutanoic(3)trifluoromethyl-cinnamic-acid(3)trifluoromethyl-nicotinic acid(21)trifluoromethylbenzoic(5)trifluoromethylbenzoic acid(1)trifluoropentanoic(4)trifluorophenylacetic(15)trifluorophenylacetic acid(6)trifluoropropanoic(4)triiodobenzoic(4)trimethoxybenzoic(2)trimethylbenzoic(15)trimethylbenzoic acid(1)
Chlorides(77683)
(2,3-dichlorophenyl)boronic acid(6)(2,6-dichlorophenyl)boronic acid(5)(2-chloroethoxy)benzene(4)(2-chlorophenyl)(phenyl)methanone(17)(2-chlorophenyl)hydrazine(13)(2-chlorophenyl)methanamine(20)(2-chlorophenyl)methanol(34)(2-chloropyridin-4-yl)boronic acid(6)(3,4-dichlorophenyl)boronic acid(6)(3-chloro-4-fluorophenyl)boronic acid(7)(3-chlorophenyl)(phenyl)methanone(10)(3-chlorophenyl)boronic acid(56)(3-chlorophenyl)hydrazine(7)(3-chlorophenyl)methanamine(12)(3-chlorophenyl)methanol(21)(3-chloropyridin-2-yl)methanamine(6)(4-chlorophenyl)(phenyl)methanone(11)(5-chloropyridin-2-yl)methanamine(5)(chloromethanetriyl)tribenzene(7)(chloromethylene)dibenzene(5)1,2,3,4-tetrachlorobenzene(7)1,2,4-trichlorobenzene(18)1,2-dibromo-3-chlorobenzene(3)1,2-dichloro-3-fluorobenzene(8)1,2-dichloro-3-methoxybenzene(4)1,2-dichloro-3-methylbenzene(6)1,2-dichloro-4-fluorobenzene(8)1,2-dichloro-4-methoxybenzene(8)1,2-dichloro-4-nitrobenzene(12)1,3-dibromo-2-chlorobenzene(5)1,3-dichloro-2-fluorobenzene(17)1,3-dichloro-2-iodobenzene(5)1,3-dichloro-2-methoxybenzene(7)1,3-dichloro-2-nitrobenzene(5)1,3-dichloroisoquinoline(14)1,4-dichloro-2-fluorobenzene(8)1,4-dichloro-2-methoxybenzene(6)1-(2-chlorophenyl)ethan-1-one(35)1-(2-chlorophenyl)piperazine(4)1-(3-chlorophenyl)-1H-pyrazole(3)1-(3-chlorophenyl)-N-methylmethanamine(7)1-(3-chlorophenyl)ethan-1-one(20)1-(3-chlorophenyl)pyrrolidine(3)1-(benzyloxy)-2-chlorobenzene(16)1-(benzyloxy)-3-chlorobenzene(6)1-(bromomethyl)-2-chlorobenzene(28)1-(bromomethyl)-3-chlorobenzene(18)1-(chloromethyl)-2-methylbenzene(6)1-benzyl-2-chlorobenzene(8)1-benzyl-3-chlorobenzene(2)1-benzyl-4-chlorobenzene(6)1-bromo-2,3-dichlorobenzene(16)1-bromo-2-chloro-3-methylbenzene(7)1-bromo-3-chloro-2-fluorobenzene(8)1-bromo-3-chloro-2-methylbenzene(3)1-bromo-4-chloro-2-fluorobenzene(14)1-chloro-2,3-difluorobenzene(25)1-chloro-2,4-difluorobenzene(19)1-chloro-2,4-dimethoxybenzene(4)1-chloro-2-(chloromethyl)benzene(12)1-chloro-2-(trifluoromethoxy)benzene(18)1-chloro-2-(trifluoromethyl)benzene(37)1-chloro-2-cyclopropylbenzene(7)1-chloro-2-ethoxybenzene(8)1-chloro-2-fluoro-3-iodobenzene(5)1-chloro-2-fluoro-4-nitrobenzene(6)1-chloro-2-iodobenzene(51)1-chloro-2-isocyanatobenzene(6)1-chloro-2-isothiocyanatobenzene(7)1-chloro-2-methyl-3-nitrobenzene(5)1-chloro-2-nitrobenzene(60)1-chloro-2-phenoxybenzene(10)1-chloro-2-propoxybenzene(4)1-chloro-3,5-dimethylbenzene(7)1-chloro-3-(chloromethyl)benzene(5)1-chloro-3-(trifluoromethoxy)benzene(10)1-chloro-3-(trifluoromethyl)benzene(60)1-chloro-3-fluoro-2-methylbenzene(5)1-chloro-3-fluoro-2-nitrobenzene(5)1-chloro-3-iodo-2-methylbenzene(3)1-chloro-3-iodobenzene(46)1-chloro-3-methoxybenzene(65)1-chloro-3-nitrobenzene(66)1-chloro-3-phenoxybenzene(5)1-chloro-4-(trifluoromethoxy)benzene(7)1-chloro-4-iodobenzene(18)1-chloro-4-methoxybenzene(50)1-chloro-4-phenoxybenzene(13)1-chloroanthracene-9,10-dione(6)1-chloroisoquinoline(20)1-chloronaphthalene(24)2,3,5-trichloropyridine(13)2,3,6-trichloropyridine(16)2,3-dichloroaniline(16)2,3-dichlorobenzaldehyde(6)2,3-dichlorophenol(6)2,3-dichloropyridine(38)2,4,5-trichloropyrimidine(6)2,4,6-trichloropyridine(5)2,4,6-trichloropyrimidine(14)2,4-dibromo-1-chlorobenzene(7)2,4-dichloro-1-fluorobenzene(18)2,4-dichloro-1-methoxybenzene(11)2,4-dichloro-1-nitrobenzene(15)2,4-dichloro-6-methylpyridine(8)2,4-dichloro-6-methylpyrimidine(7)2,4-dichlorobenzaldehyde(6)2,4-dichlorobenzonitrile(4)2,4-dichloropyridine(50)2,4-dichloroquinazoline(19)2,4-dichlorothiazole(4)2,5-dichlorophenol(5)2,5-dichloropyrazine(4)2,5-dichloropyridine(32)2,6-dichloro-3-fluoropyridine(10)2,6-dichloro-3-methylpyridine(5)2,6-dichloro-3-nitropyridine(6)2,6-dichloro-4-methylpyridine(5)2,6-dichloroaniline(23)2,6-dichlorobenzoic acid(9)2,6-dichlorobenzonitrile(8)2,6-dichloronicotinonitrile(8)2,6-dichlorophenol(12)2,6-dichloropyrazine(14)2,6-dichloropyridin-3-amine(11)2,6-dichloropyridine(49)2-(2-chlorophenyl)acetic acid(19)2-(2-chlorophenyl)acetonitrile(15)2-(2-chlorophenyl)ethan-1-ol(6)2-(2-chlorophenyl)pyridine(3)2-(2-chlorophenyl)pyrrolidine(17)2-(3-chlorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(8)2-(3-chlorophenyl)acetonitrile(7)2-(4-chlorophenoxy)acetic acid(2)2-(chloromethyl)-1H-benzo[d]imidazole(9)2-(chloromethyl)-3-methylpyridine(12)2-(chloromethyl)furan(3)2-(chloromethyl)pyridine(12)2-(chloromethyl)pyridine 1-oxide(4)2-(chloromethyl)pyrimidine(5)2-(chloromethyl)thiazole(5)2-amino-3-chlorobenzoic acid(4)2-amino-3-chlorobenzonitrile(3)2-amino-6-chlorophenol(4)2-bromo-1,3-dichlorobenzene(11)2-bromo-1,4-dichlorobenzene(9)2-bromo-1-chloro-4-methylbenzene(6)2-bromo-4-chloro-1-iodobenzene(5)2-bromo-4-chloro-1-methoxybenzene(4)2-bromo-4-chloro-1-methylbenzene(5)2-bromo-4-chloroaniline(12)2-bromo-4-chlorophenol(8)2-bromo-6-chloroaniline(10)2-bromo-6-chlorophenol(4)2-chloro-1,1'-biphenyl(8)2-chloro-1,3,5-triazine(10)2-chloro-1,3-difluorobenzene(22)2-chloro-1,3-dimethoxybenzene(3)2-chloro-1,3-dinitrobenzene(2)2-chloro-1,4-difluorobenzene(17)2-chloro-1,4-dimethoxybenzene(3)2-chloro-1-fluoro-4-nitrobenzene(11)2-chloro-1-methyl-3-nitrobenzene(3)2-chloro-1-phenylethan-1-one(15)2-chloro-1H-benzo[d]imidazole(17)2-chloro-3,6-dimethylpyridine(4)2-chloro-3-(trifluoromethyl)aniline(3)2-chloro-3-(trifluoromethyl)pyridine(12)2-chloro-3-fluoropyridin-4-amine(3)2-chloro-3-hydroxybenzaldehyde(4)2-chloro-3-methoxypyridine(12)2-chloro-3-methylaniline(3)2-chloro-3-methylbenzoic acid(4)2-chloro-3-methylbenzonitrile(4)2-chloro-3-methylpyrazine(7)2-chloro-3-methylpyridine(17)2-chloro-3-nitropyridine(10)2-chloro-4,6-dimethylpyridine(7)2-chloro-4-(trifluoromethyl)pyridine(9)2-chloro-4-ethoxypyridine(5)2-chloro-4-fluoro-1-nitrobenzene(9)2-chloro-4-fluoroaniline(10)2-chloro-4-fluoropyridine(10)2-chloro-4-methoxypyridine(22)2-chloro-4-methyl-1-nitrobenzene(6)2-chloro-4-methylaniline(5)2-chloro-4-methylbenzoic acid(3)2-chloro-4-methylpyridine(31)2-chloro-4-methylpyrimidine(16)2-chloro-4-methylthiazole(5)2-chloro-4-phenylpyrimidine(7)2-chloro-5-(trifluoromethyl)pyridine(4)2-chloro-5-fluoropyridin-4-amine(3)2-chloro-5-fluoropyridine(16)2-chloro-5-methoxyaniline(5)2-chloro-5-methylpyridine(11)2-chloro-5-methylpyrimidine(5)2-chloro-5-methylthiazole(5)2-chloro-5-nitropyridine(8)2-chloro-6-(trifluoromethyl)aniline(4)2-chloro-6-(trifluoromethyl)pyridine(16)2-chloro-6-fluoroaniline(6)2-chloro-6-fluorophenol(4)2-chloro-6-fluoropyridine(14)2-chloro-6-iodophenol(3)2-chloro-6-methoxyaniline(3)2-chloro-6-methoxyphenol(4)2-chloro-6-methoxypyridine(24)2-chloro-6-methyl-3-nitropyridine(4)2-chloro-6-methylaniline(10)2-chloro-6-methylnicotinonitrile(10)2-chloro-6-methylphenol(4)2-chloro-6-methylpyrazine(12)2-chloro-6-methylpyridine(49)2-chloro-6-nitroaniline(10)2-chloro-7H-purine(3)2-chloro-7H-pyrrolo[2,3-d]pyrimidine(13)2-chloro-9H-purine(5)2-chloro-N-phenylacetamide(21)2-chloro-N-phenylaniline(8)2-chloroaniline(96)2-chlorobenzamide(12)2-chlorobenzene-1,3-diamine(3)2-chlorobenzene-1,3-diol(4)2-chlorobenzene-1,4-diamine(4)2-chlorobenzenesulfonyl chloride(24)2-chlorobenzenethiol(7)2-chlorobenzo[d]oxazole(11)2-chlorobenzo[d]thiazole(21)2-chlorobenzoic acid(57)2-chlorobenzonitrile(35)2-chlorobenzoyl chloride(13)2-chlorocyclohexa-2,5-diene-1,4-dione(4)2-chlorofuran(3)2-chloroisonicotinaldehyde(14)2-chloroisonicotinic acid(17)2-chloroisonicotinonitrile(7)2-chloronaphthalene(6)2-chloronicotinic acid(7)2-chloronicotinonitrile(9)2-chlorooxazole(7)2-chlorophenol(68)2-chloropyrazine(24)2-chloropyridin-3-amine(11)2-chloropyridin-3-ol(9)2-chloropyridin-4-amine(28)2-chloropyridine 1-oxide(10)2-chloroquinoxaline(39)2-chlorothiazole(32)2-chlorothiophene(20)3,4-dichloroaniline(13)3,5-dichloropyridine(16)3,6-dichloropyridazine(20)3-(3-chlorophenyl)propanoic acid(4)3-(chloromethyl)pyridine(22)3-chloro-1,1'-biphenyl(12)3-chloro-1-methyl-1H-pyrazole(7)3-chloro-1-phenylpropan-1-one(7)3-chloro-1H-indazole(13)3-chloro-2-fluoroaniline(8)3-chloro-2-fluorobenzaldehyde(8)3-chloro-2-fluorobenzoic acid(7)3-chloro-2-fluorobenzonitrile(4)3-chloro-2-fluoropyridine(13)3-chloro-2-hydroxybenzaldehyde(4)3-chloro-2-methoxypyridine(16)3-chloro-2-methylaniline(10)3-chloro-2-methylpyridine(4)3-chloro-2-nitroaniline(4)3-chloro-4-(trifluoromethyl)aniline(5)3-chloro-4-fluoroaniline(14)3-chloro-4-fluorobenzoic acid(6)3-chloro-4-hydroxybenzaldehyde(4)3-chloro-4-methylaniline(8)3-chloro-4-methylpyridazine(7)3-chloro-4-nitroaniline(4)3-chloro-5-fluoropyridine(10)3-chloro-5-methylpyridine(7)3-chloro-5-nitropyridine(5)3-chloro-9H-carbazole(3)3-chlorobenzaldehyde(53)3-chlorobenzene-1,2-diamine(4)3-chlorobenzene-1,2-diol(6)3-chlorobenzenesulfonyl chloride(21)3-chlorobenzo[b]thiophene(6)3-chlorobenzoic acid(85)3-chlorobenzonitrile(63)3-chloroimidazo[1,2-a]pyridine(3)3-chloroisoquinoline(20)3-chlorophenol(58)3-chloropicolinic acid(9)3-chloropicolinonitrile(6)3-chloropyridazine(44)3-chloropyridin-2-amine(17)3-chloropyridin-2-ol(9)3-chlorothiophene(17)4,5-dichloropyrimidine(7)4,6-dichloro-2-methylpyrimidine(7)4,6-dichloropyridin-2-amine(6)4,6-dichloropyridin-3-amine(4)4,6-dichloropyrimidine(14)4-chloro-1,5-naphthyridine(6)4-chloro-1-phenylbutan-1-one(7)4-chloro-1H-indazole(18)4-chloro-1H-indole(22)4-chloro-1H-pyrazole(10)4-chloro-1H-pyrazolo[3,4-b]pyridine(5)4-chloro-1H-pyrazolo[3,4-d]pyrimidine(5)4-chloro-1H-pyrazolo[4,3-c]pyridine(7)4-chloro-1H-pyrrolo[3,2-c]pyridine(10)4-chloro-2,6-dimethylpyridine(4)4-chloro-2-methoxypyridine(6)4-chloro-2-phenylpyrimidine(5)4-chloro-3-fluoroaniline(8)4-chloro-3-methylaniline(8)4-chloro-3-methylpyridine(7)4-chloro-5-methylpyrimidine(8)4-chloro-N-phenylaniline(6)4-chlorobenzene-1,3-diol(3)4-chlorobenzenesulfonic acid(3)4-chlorobenzenesulfonyl chloride(12)4-chlorobenzo[d]thiazole(7)4-chloroindoline-2,3-dione(6)4-chloroisoquinoline(11)4-chloronicotinic acid(4)4-chloropyridazine(16)4-chloropyridin-2-amine(6)4-chloropyridine(31)4-chloroquinazoline(39)5,6-dichloro-1H-benzo[d]imidazole(4)5,6-dichloropyridin-2-amine(4)5-bromo-4-chloropyrimidine(4)5-chloro-1,2,3,4-tetrahydroisoquinoline(3)5-chloro-1H-imidazole(2)5-chloro-1H-indazole(9)5-chloro-1H-indole(29)5-chloro-1H-indole-2-carboxylic acid(6)5-chloro-1H-pyrazole(8)5-chloro-1H-pyrazolo[4,3-b]pyridine(6)5-chloro-1H-pyrrolo[2,3-b]pyridine(13)5-chloro-1H-pyrrolo[3,2-b]pyridine(6)5-chloro-2,3-dihydrobenzofuran(5)5-chloro-2-fluoropyridine(13)5-chloro-2-methoxypyridine(10)5-chloro-2-methylpyridine(10)5-chlorobenzo[b]thiophene(4)5-chlorobenzo[d]oxazole(5)5-chlorobenzo[d]thiazole(5)5-chloroimidazo[1,2-a]pyridine(3)5-chloroindolin-2-one(3)5-chloroisoquinoline(6)5-chloropicolinic acid(7)5-chloropicolinonitrile(6)5-chloropyrazolo[1,5-a]pyrimidine(24)5-chloropyridin-2-amine(26)5-chloropyridin-2-ol(12)5-chloropyridin-3-amine(7)5-chloropyrimidine(8)5-chloroquinazoline(4)6-chloro-1,2,3,4-tetrahydroisoquinoline(3)6-chloro-1,2,3,4-tetrahydroquinoline(3)6-chloro-1H-benzo[d]imidazole(11)6-chloro-1H-indazole(14)6-chloro-1H-indole(19)6-chloro-1H-pyrazolo[4,3-c]pyridine(8)6-chloro-1H-pyrrolo[2,3-b]pyridine(13)6-chloro-1H-pyrrolo[3,2-c]pyridine(13)6-chloro-2,3-dihydrobenzofuran(3)6-chloro-3,4-dihydro-2H-benzo[b][1,4]oxazine(5)6-chloro-3-nitropyridin-2-amine(4)6-chloro-5-(trifluoromethyl)pyridin-2-amine(3)6-chlorobenzo[d]oxazole(4)6-chlorobenzo[d]thiazole(6)6-chlorochroman-4-one(6)6-chlorochromane(6)6-chloroindoline(3)6-chloroindoline-2,3-dione(5)6-chloronicotinaldehyde(12)6-chloronicotinic acid(4)6-chloropicolinic acid(12)6-chloropicolinonitrile(9)6-chloropyridazin-3(2H)-one(5)6-chloropyridin-2-amine(21)6-chloropyridin-2-ol(8)6-chloropyridin-3-amine(4)6-chloropyridine-3-sulfonyl chloride(5)6-chloroquinoxaline(3)7-chloro-1,2,3,4-tetrahydroisoquinoline(5)7-chloro-1H-indazole(7)7-chloro-1H-indole(21)7-chloro-1H-pyrrolo[2,3-c]pyridine(5)7-chloro-2,3-dihydrobenzofuran(3)7-chloro-3,4-dihydronaphthalen-1(2H)-one(6)7-chlorobenzo[d]thiazole(6)7-chlorochromane(5)7-chloroisoquinoline(6)7-chloropyrazolo[1,5-a]pyrimidine(8)8-chlorochromane(5)8-chloroimidazo[1,2-a]pyridine(7)8-chloroisoquinoline(7)8-chloroquinazoline(5)ethyl 4,6-dichloronicotinate(4)ethyl 5-chloro-1H-indole-2-carboxylate(5)ethyl 6-chloropicolinate(7)ethyl 7-chloro-1H-indole-2-carboxylate(5)methyl 2,6-dichlorobenzoate(6)methyl 2-chloro-4-fluorobenzoate(5)methyl 2-chloro-6-fluorobenzoate(7)methyl 2-chlorobenzoate(33)methyl 2-chloroisonicotinate(11)methyl 2-chloronicotinate(6)methyl 3-chlorobenzoate(58)methyl 3-chloropicolinate(9)methyl 6-chloropicolinate(11)N-(2-chlorophenyl)acetamide(13)N-(3-chlorophenyl)benzenesulfonamide(5)N-(3-chlorophenyl)quinazolin-4-amine(10)N-(4-chlorophenyl)acetamide(6)phenyl carbonochloridate(5)pyridine-2-sulfonyl chloride(9)pyridine-3-sulfonyl chloride(25)tert-butyl (2-chloropyridin-4-yl)carbamate(4)2,2,2-trichloro-ethanone(5)2-chloro-N-acetamide(44)2-chloro-N-methyl-acetamide(4)2-chloro-N-methylacetamide(4)2-chloroacetyl(39)2-chloroethyl(25)3-chloro-1-propanone(9)3-chloro-propanamide(4)3-chloropropyl(14)4-chloro-butan-1-one(5)4-chlorobutyl(4)benzothiazole-chloride(41)benzoxazole-chloride(4)bromo-chloro-benzene(25)carbonochloridate(6)chlorindole-carboxylic acid(27)chlorindole-carboxylic-acid(2)chloro-benzimidazole(5)chloro-carbazole(20)chloro-fluoro-benzene(69)chloro-imidazopyridine(78)chloro-indazole(44)chloro-indazole-carboxylate(3)chloro-inden-amine(10)chloro-indene(47)chloro-indole-carboxylate(41)chloro-iodo-trifluoromethyl-pyridine(7)chloro-methyl indole(111)chloro-methyl-indazole(27)chloro-methyl-pyrazole(155)chloro-naphthyridin-one(5)chloro-naphthyridine(76)chloro-nitrobenzene(40)chloro-one(551)chloro-phenyl-boronic-acid(49)chloro-propylamine-hydrochloride(2)chloro-purine(34)chloro-pyrazolamide(1)chloro-pyrazole(23)chloro-pyrazole-carboxylate(36)chloro-pyrazolopyridine(38)chloro-pyrrolopyridine(33)chloro-pyrrolopyrimidine(102)chloro-thiadiazole(29)chloro-trifluoromethyl-pyridin-amine(28)chloro-trifluoromethyl-pyridine(145)chloroacetamide(17)chloroacetyl(12)chloroaniline(18)chlorobenzaldehyde(18)chlorobenzamide(12)chlorobenzene(107)chlorobenzenesulfonamide(62)chlorobenzofuran(10)chlorobenzothiazole(4)chlorobenzotrifluoride(3)chlorobenzyl(75)chlorobutyl(6)chlorobutyl(6)chlorocarbonyl(7)chlorochroman(11)chloroethoxy(10)chloroethoxy(10)chloroethyl(89)chloroethylamino(8)chloroimidazo(62)chloroindole(6)chloroisobenzofuran(10)chloromethyl(479)chlorooxazole(5)chlorophenyl(925)chlorophthalazin(16)chloropicolinaldehyde(8)chloropicolinamide(5)chloropropane(18)chloropropane(18)chloropropoxy(2)chloropropoxy(2)chloropropyl(23)chloropyrazine(43)chloropyridazine(33)chloropyridine(254)chloropyrimidine(86)chloroquinazoline(30)chloroquinoline(138)chloroquinoxaline(14)chlorotoluene(4)dichloride(28)dichloro(5)dichloroaniline(9)dichloroanthracene(3)dichlorobenzaldehyde(13)dichlorobenzamide(8)dichlorobenzenesulfonamide(18)dichloroisonicotinaldehyde(4)dichloromethyl(3)dichloromethyl(3)dichloronicotinaldehyde(6)dichloronicotinate(12)dichloronicotinic(7)dichloronicotinic(7)dichloronicotinic acid(3)dichloropalladium(4)dichloropalladium(4)dichloropyridazine(17)dichloropyrimidine(38)dichloroquinoline(16)nicotinoyl-chloride(7)tetrachloro(2)tetrachloro(2)tetrahydroisoquinoline-chloride(44)tetrahydroquinoline-chloride(7)trichloride(2)trichloro(36)trichloro(36)trichloroacetimidate(5)trichloroaniline(6)trichlorobenzaldehyde(4)trichloroethyl(2)trichloroethyl(2)trichloromethyl(11)trichloromethyl(11)
Difluoromethyls(32559)
(difluoromethyl)benzene(48)1-(difluoromethoxy)-2-fluorobenzene(9)1-(difluoromethyl)-2-fluorobenzene(9)1-bromo-2-(difluoromethyl)benzene(5)2-(difluoromethoxy)phenol(5)2-(difluoromethyl)pyridine(21)3-(difluoromethyl)pyridine(5)4-(difluoromethoxy)aniline(5)5-(difluoromethyl)-1H-pyrazole(6)difluoromethyl-benzene(6)difluoromethyl-pyrazole(30)difluoromethyl-pyrazole-carboxylate(5)difluoromethyl-pyridine(4)other-difluoromethyl(32)
Esters(57023)
(2R,3S)-2-((benzoyloxy)methyl)tetrahydrofuran-3-yl benzoate(8)(9H-fluoren-9-yl)methyl (3-phenylpropyl)carbamate(24)1,3-dioxoisoindolin-2-yl 2-phenylacetate(5)1-benzyl 2-methyl pyrrolidine-1,2-dicarboxylate(7)1-benzyl 3-methyl piperidine-1,3-dicarboxylate(3)1-benzyl 4-(tert-butyl) piperazine-1,4-dicarboxylate(10)1H-indol-3-yl acetate(5)2,5-dioxopyrrolidin-1-yl benzoate(8)2-((tert-butoxycarbonyl)amino)benzoic acid(8)2-(diethylamino)ethyl benzoate(3)2-ethylhexyl benzoate(13)2-hydroxyethyl benzoate(3)2-phenoxyacetyl chloride(3)4-cyanophenyl benzoate(11)allyl benzoate(4)azetidin-3-yl acetate(3)benzoic anhydride(6)benzyl acetate(7)benzyl benzoate(12)benzyl benzylcarbamate(16)benzyl cyclohexylcarbamate(9)benzyl morpholine-4-carboxylate(5)benzyl phenylcarbamate(15)benzyl piperidine-1-carboxylate(50)butyl benzoate(9)dimethyl isophthalate(16)dimethyl phthalate(8)dimethyl pyridine-2,6-dicarboxylate(7)ethane-1,2-diyl dibenzoate(11)ethyl 1-phenyl-1H-pyrazole-3-carboxylate(4)ethyl 1-phenyl-1H-pyrazole-4-carboxylate(13)ethyl 1-phenylcyclopropane-1-carboxylate(3)ethyl 1H-1,2,4-triazole-3-carboxylate(3)ethyl 1H-imidazole-2-carboxylate(6)ethyl 1H-indole-3-carboxylate(6)ethyl 1H-pyrazole-4-carboxylate(11)ethyl 1H-pyrrole-2-carboxylate(24)ethyl 1H-pyrrole-3-carboxylate(8)ethyl 2-(imidazo[1,2-a]pyridin-3-yl)acetate(3)ethyl 2-(pyridin-2-yl)acetate(11)ethyl 2-aminobenzoate(13)ethyl 2-aminothiazole-4-carboxylate(6)ethyl 2-aminothiazole-5-carboxylate(5)ethyl 2-bromo-2-phenylacetate(6)ethyl 2-fluorobenzoate(26)ethyl 2-hydroxybenzoate(11)ethyl 2-iodobenzoate(4)ethyl 2-methylbenzoate(15)ethyl 2-methylnicotinate(6)ethyl 2-nitrobenzoate(7)ethyl 2-oxo-2-(phenylamino)acetate(6)ethyl 2-oxo-2-phenylacetate(10)ethyl 2-oxo-2H-chromene-3-carboxylate(5)ethyl 2-phenoxyacetate(8)ethyl 2-phenylacetate(37)ethyl 2-phenylcyclopropane-1-carboxylate(5)ethyl 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate(6)ethyl 3-aminobenzoate(14)ethyl 3-fluorobenzoate(17)ethyl 3-hydroxybenzoate(7)ethyl 3-iodobenzoate(8)ethyl 3-nitrobenzoate(14)ethyl 3-oxo-3-phenylpropanoate(24)ethyl 3-phenylpropanoate(10)ethyl 4-aminobenzoate(12)ethyl 4-chloropyrimidine-5-carboxylate(5)ethyl 5-fluoro-1H-indole-2-carboxylate(5)ethyl 6-chloro-1H-indole-2-carboxylate(5)ethyl benzo[b]thiophene-2-carboxylate(9)ethyl cinnamate(18)ethyl cyclohex-1-ene-1-carboxylate(5)ethyl cyclohexanecarboxylate(19)ethyl cyclopropanecarboxylate(8)ethyl furan-2-carboxylate(6)ethyl imidazo[1,2-a]pyridine-2-carboxylate(25)ethyl imidazo[1,2-a]pyridine-3-carboxylate(13)ethyl isonicotinate(12)ethyl isoxazole-3-carboxylate(8)ethyl isoxazole-4-carboxylate(5)ethyl morpholine-2-carboxylate(4)ethyl nicotinate(25)ethyl oxazole-4-carboxylate(8)ethyl oxazole-5-carboxylate(3)ethyl oxirane-2-carboxylate(4)ethyl picolinate(41)ethyl piperidine-2-carboxylate(9)ethyl piperidine-3-carboxylate(13)ethyl piperidine-4-carboxylate(9)ethyl pyrazine-2-carboxylate(10)ethyl pyridazine-3-carboxylate(6)ethyl pyrimidine-4-carboxylate(7)ethyl pyrimidine-5-carboxylate(5)ethyl quinazoline-2-carboxylate(4)ethyl thiazole-2-carboxylate(7)ethyl thiazole-4-carboxylate(7)ethyl thiazole-5-carboxylate(7)ethyl thiophene-2-carboxylate(19)ethyl thiophene-3-carboxylate(11)hexyl benzoate(5)isobutyl benzoate(9)isopropyl benzoate(10)methyl 1-naphthoate(7)methyl 1H-benzo[d]imidazole-2-carboxylate(3)methyl 1H-imidazole-2-carboxylate(4)methyl 1H-imidazole-5-carboxylate(5)methyl 1H-indazole-3-carboxylate(23)methyl 1H-indazole-4-carboxylate(10)methyl 1H-indazole-5-carboxylate(3)methyl 1H-indazole-6-carboxylate(9)methyl 1H-indazole-7-carboxylate(8)methyl 1H-indole-3-carboxylate(21)methyl 1H-indole-4-carboxylate(9)methyl 1H-indole-5-carboxylate(7)methyl 1H-indole-6-carboxylate(9)methyl 1H-indole-7-carboxylate(7)methyl 1H-pyrazole-4-carboxylate(9)methyl 1H-pyrrole-2-carboxylate(22)methyl 1H-pyrrole-3-carboxylate(5)methyl 1H-pyrrolo[2,3-b]pyridine-2-carboxylate(3)methyl 1H-pyrrolo[2,3-b]pyridine-3-carboxylate(4)methyl 1H-pyrrolo[2,3-b]pyridine-6-carboxylate(3)methyl 2,6-dichlorobenzoate(6)methyl 2,6-difluorobenzoate(13)methyl 2-(1H-indol-3-yl)acetate(3)methyl 2-(2-bromophenyl)acetate(7)methyl 2-(4-hydroxyphenyl)acetate(6)methyl 2-(bromomethyl)benzoate(26)methyl 2-(chlorosulfonyl)benzoate(7)methyl 2-(morpholin-3-yl)acetate(3)methyl 2-(pyridin-2-yl)acetate(9)methyl 2-(trifluoromethyl)benzoate(12)methyl 2-amino-2-phenylacetate(26)methyl 2-aminobenzoate(52)methyl 2-aminothiazole-4-carboxylate(6)methyl 2-bromo-2-phenylacetate(7)methyl 2-bromo-3-nitrobenzoate(2)methyl 2-bromo-5-fluorobenzoate(4)methyl 2-bromo-6-fluorobenzoate(6)methyl 2-bromo-6-methylbenzoate(6)methyl 2-bromobenzoate(44)methyl 2-chloro-4-fluorobenzoate(5)methyl 2-chloro-6-fluorobenzoate(7)methyl 2-chlorobenzoate(33)methyl 2-cyanobenzoate(10)methyl 2-fluorobenzoate(46)methyl 2-hydroxybenzoate(39)methyl 2-iodobenzoate(24)methyl 2-methoxybenzoate(44)methyl 2-methyl-2H-indazole-3-carboxylate(3)methyl 2-methylbenzoate(51)methyl 2-naphthoate(11)methyl 2-nitrobenzoate(36)methyl 2-phenoxyacetate(9)methyl 2-phenoxybenzoate(4)methyl 2-phenylacetate(63)methyl 3-(benzyloxy)benzoate(5)methyl 3-(bromomethyl)benzoate(14)methyl 3-(trifluoromethyl)benzoate(10)methyl 3-amino-3-phenylpropanoate(19)methyl 3-aminobenzoate(49)methyl 3-bromo-4-chlorobenzoate(5)methyl 3-bromo-4-fluorobenzoate(6)methyl 3-bromo-4-methoxybenzoate(8)methyl 3-bromo-4-methylbenzoate(5)methyl 3-bromobenzoate(60)methyl 3-bromopyrazine-2-carboxylate(5)methyl 3-chlorobenzoate(58)methyl 3-cyanobenzoate(14)methyl 3-fluorobenzoate(80)methyl 3-formylbenzoate(7)methyl 3-hydroxybenzoate(31)methyl 3-iodo-4-methylbenzoate(6)methyl 3-iodobenzoate(21)methyl 3-methoxybenzoate(36)methyl 3-methylbenzoate(37)methyl 3-nitrobenzoate(51)methyl 3-oxo-3-phenylpropanoate(8)methyl 3-phenylpropanoate(22)methyl 4-amino-3-bromobenzoate(5)methyl 4-amino-3-iodobenzoate(5)methyl 4-amino-3-nitrobenzoate(3)methyl 4-aminobenzoate(32)methyl 4-fluorobenzoate(44)methyl 4-nitrobenzoate(15)methyl 4-oxocyclohexane-1-carboxylate(4)methyl 4-oxopiperidine-2-carboxylate(3)methyl 4-phenoxybenzoate(3)methyl 5-amino-2-bromobenzoate(4)methyl 5-hydroxypicolinate(5)methyl 5-oxopyrrolidine-2-carboxylate(3)methyl 5-phenylisoxazole-3-carboxylate(6)methyl 6-bromopyrazine-2-carboxylate(7)methyl 6-chloropyrazine-2-carboxylate(5)methyl 6-methoxynicotinate(4)methyl [1,1'-biphenyl]-4-carboxylate(8)methyl azetidine-2-carboxylate(3)methyl benzo[b]thiophene-2-carboxylate(27)methyl benzo[d]oxazole-2-carboxylate(3)methyl benzo[d]thiazole-6-carboxylate(5)methyl bicyclo[1.1.1]pentane-1-carboxylate(9)methyl cinnamate(21)methyl cyclobutanecarboxylate(19)methyl cyclohex-3-ene-1-carboxylate(2)methyl cyclohexanecarboxylate(26)methyl cyclopentanecarboxylate(16)methyl cyclopropanecarboxylate(8)methyl furan-2-carboxylate(14)methyl imidazo[1,2-a]pyridine-8-carboxylate(3)methyl indoline-6-carboxylate(3)methyl isonicotinate(16)methyl isoquinoline-4-carboxylate(6)methyl L-phenylalaninate(17)methyl morpholine-2-carboxylate(4)methyl morpholine-3-carboxylate(10)methyl nicotinate(52)methyl oxazole-4-carboxylate(9)methyl piperidine-2-carboxylate(12)methyl piperidine-3-carboxylate(19)methyl prolinate(43)methyl pyrazine-2-carboxylate(19)methyl pyridazine-3-carboxylate(7)methyl pyrimidine-2-carboxylate(10)methyl pyrimidine-4-carboxylate(11)methyl pyrimidine-5-carboxylate(5)methyl pyrrolidine-3-carboxylate(9)methyl thiazole-2-carboxylate(6)methyl thiazole-4-carboxylate(6)methyl thiophene-3-carboxylate(33)phenyl (1r,4r)-[1,1'-bi(cyclohexane)]-4-carboxylate(6)phenyl dimethylcarbamate(5)phenyl ethyl(methyl)carbamate(5)propyl benzoate(9)tert-butyl (1-phenylethyl)carbamate(14)tert-butyl (2,3-dihydro-1H-inden-1-yl)carbamate(3)tert-butyl (2-phenylcyclopropyl)carbamate(3)tert-butyl (3-oxocyclohexyl)carbamate(3)tert-butyl (morpholin-2-ylmethyl)carbamate(3)tert-butyl (tetrahydrofuran-3-yl)carbamate(4)tert-butyl 1H-benzo[d]imidazole-1-carboxylate(5)tert-butyl 1H-imidazole-1-carboxylate(6)tert-butyl 1H-indazole-1-carboxylate(29)tert-butyl 1H-indole-1-carboxylate(18)tert-butyl 1H-pyrrole-1-carboxylate(8)tert-butyl 1H-pyrrole-2-carboxylate(3)tert-butyl 1H-pyrrolo[2,3-b]pyridine-1-carboxylate(9)tert-butyl 3,4-dihydroisoquinoline-2(1H)-carboxylate(19)tert-butyl 3-((methylsulfonyl)oxy)pyrrolidine-1-carboxylate(5)tert-butyl 3-oxopiperazine-1-carboxylate(7)tert-butyl 4-phenylpiperazine-1-carboxylate(23)tert-butyl 4-phenylpiperidine-1-carboxylate(16)tert-butyl azetidine-1-carboxylate(64)tert-butyl benzoate(44)tert-butyl benzylcarbamate(42)tert-butyl bicyclo[1.1.1]pentan-1-ylcarbamate(5)tert-butyl cyclopentylcarbamate(28)tert-butyl isoindoline-2-carboxylate(13)tert-butyl methyl(phenyl)carbamate(8)tert-butyl morpholine-4-carboxylate(23)tert-butyl o-tolylcarbamate(7)tert-butyl phenethylcarbamate(9)tert-butyl piperazine-1-carboxylate(30)tert-butyl pyrrolidine-1-carboxylate(21)(2-hydroxyethoxy)carbonyl(3)(R)-methyl 2-amino-acetate(11)(R)-methyl 2-amino-propanoate(14)(S)-methyl 2-((tert-butoxycarbonyl)amino)-propanoate(12)(S)-methyl 2-amino-2-acetate(8)2-ethylhexyl carboxylate(13)6-oxy-hexyl acrylate(5)acetate(183)acetoxybenzoic(4)acetoxymethyl(85)acetoxyphenyl(1)acetyloxy(54)acrylate(118)acryloyloxy(3)adipate(16)adipate(16)allyl carboxylate(8)amino-carboxylate(969)aminobenzoate(18)aminobenzylcarbamate(8)aminobutanoate(14)aminocyclobutanecarboxylate(8)aminocyclobutanecarboxylic(6)aminocyclobutanecarboxylic acid(4)aminocyclohexanecarboxylate(20)aminocyclohexanecarboxylic(16)aminocyclohexanecarboxylic acid(3)aminocyclopentanecarboxylate(10)aminocyclopentanecarboxylic(13)aminocyclopropanecarboxylate(4)aminoheptanoate(9)aminohexanoate(6)aminoimidazole-carboxylate(23)aminoisonicotinate(4)aminoisophthalate(5)aminonicotinate(7)aminopentanedioate(15)aminopentanoate(7)aminopropanoate(16)aminosuccinate(4)azanediyldiacetate(1)azanediyldiacetate(1)benzimidazole-carboxylate(6)benzoate(477)benzofuran-carboxylate(5)benzopyran-carboxylate(3)benzothiazole-carboxylate(18)benzothiophene-carboxylate(36)benzoxazine-carboxylate(9)benzoxazole-carboxylate(3)benzoyloxy(14)benzylcarbamate(8)bromo-carboxylate(41)bromo-pyrrolopyridine-carboxylate(20)bromoacetate(3)bromobenzoate(26)bromobutanoate(10)bromocyclobutanecarboxylate(6)bromocyclopropanecarboxylate(11)bromodecanoate(20)bromoheptanoate(5)bromohexanoate(9)bromoimidazole carboxylate(23)bromoisonicotinate(8)bromoisophthalate(3)bromonicotinate(13)bromopropanoate(9)bromoundecanoate(3)butanoate(34)butoxycarbonyl(1675)butoxycarbonyl(1675)butyl carboxylate(15)butylcarbamate(2)butyrate(6)carbamate(1539)carbonodithioate(6)carbothioate(2)carboxylate-hydrochloride(45)chlorobenzoate(49)chlorobenzylphosphonate(3)chloroformate(4)chloropicolinate(30)chloropropanoate(5)chroman-carboxylate(4)cyanoacrylate(6)cyanocyclopropanecarboxylate(4)cyclobutanecarboxylate(7)cyclohexanecarboxylate(5)cyclopentanecarboxylate(3)cyclopropanecarboxylate(11)decanedioate(9)decanedioate(9)decanoate(7)decanoate(7)diacetate(56)diacetoxy(1)diacrylate(16)diaminobenzoate(7)dibromobenzoate(4)dibromohexanedioate(2)dicarbamate(14)dicarboxylate(381)dichlorobenzoate(24)dichloroisonicotinate(9)dichloronicotinate(12)diethyl 1,1-dicarboxylate(7)diethyl 2-methylmalonate(4)diethyl malonate(6)diethylcarbamodithioate(1)difluorocyclobutanecarboxylate(4)dimethacrylate(1)dimethoxypropanoate(7)dimethoxypropanoate(7)dimethyl malonate(8)dimethyl-carboxylate(203)dimethyl-pyrazole-carboxylate(16)dimethylbutanoate(14)dimethylcarbamate(3)dimethylnicotinate(10)dimethylnicotinate(10)diphenylacetoxy(1)diphenylphosphinate(2)dipropanoate(6)dipropanoate(6)diyldicarbamate(5)dodecanoate(11)enecarboxylate(9)enoate(116)ethoxycarbonyl(70)ethyl 2,4-dioxobutanoate(6)ethyl 2-(amino)-2-oxoacetate(5)ethyl 2-amino-propanoate(5)ethyl 2-aminoacetate(6)ethyl 2-bromo-acetate(6)ethyl 2-methyl-propanoate(6)ethyl 2-oxoacetate(21)ethyl 2-oxyacetate(16)ethyl 3-acrylate(30)ethyl 3-amino-propanoate(5)ethyl 3-oxopropanoate(45)ethyl 3-propanoate(26)ethyl acetate(155)ethyl acrylate(5)ethyl aminocarboxylate(5)ethyl butanoate(5)ethyl carboxylate(4)ethyl cyano-carboxylate(8)ethyl methylcarboxylate(14)ethyl thioacetate(2)ethylbutanoate(6)ethynylbenzoate(7)ethynylbenzylcarbamate(3)fluoronicotinate(19)Fmoc(386)formate(4)glutarate(3)glutarate(3)heptanedioate(2)hexanoate(14)hexanoate(14)hydrazinecarboxylate(12)hydroxycyclopentanecarboxylate(4)imidazole-carboxylate(69)imidazopyridine-carboxylate(77)iminoacetate(4)indazole-carboxylate(36)indene-carboxylate(27)indolecarboxylate(2)indoline-carboxylate(59)iodoacetate(10)iodoacrylate(6)iodobutanoate(4)iodonicotinate(13)iodopropanoate(1)isoamyl benzoate(3)isobutyl-carboxylate(5)isobutyrate(18)isobutyrate(18)isopropyl carboxylate(16)isoquinolinecarboxylate(4)isoxazole-carboxylate(45)mercaptoacetate(6)mercaptopropionate(1)methacrylate(89)methacryloyloxy(7)methoxyacetate(4)methoxyacrylate(31)methoxycarbonyl(169)methoxycarbonyl(169)methoxyisonicotinate(11)methoxyisonicotinate(11)methoxynicotinate(24)methoxynicotinate(24)methoxypropanoate(5)methoxypropanoate(5)methyl ((tert-butoxycarbonyl)amino)-carboxylate(6)methyl (R)-3-amino-propanoate(9)methyl 2,4-dioxo-butanoate(7)methyl 2-amino-acetate(18)methyl 2-amino-propanoate(5)methyl 2-bromo-acetate(7)methyl 2-hydroxyacetate(5)methyl 2-methylpropanoate(8)methyl 2-oxoacetate(8)methyl 2-oxy-acetate(14)methyl 3-amino-propanoate(9)methyl 3-oxopropanoate(16)methyl 3-propanoate(38)methyl acetate(79)methyl acrylate(6)methyl aminoacetate(4)methyl aminocarboxylate(10)methyl butanoate(5)methyl carbonate(5)methyl hydroxycarboxylate(9)methyl methacrylate(5)methyl methyl-carboxylate(13)methyl propanoate(5)methyl-propanoate-hydrochloride(99)methylacrylate(13)methylbutanoate(38)methylcyclobutanecarboxylate(5)methylhexanoate(45)methylhexanoate(45)methylisonicotinate(14)methylmalonate(6)methylnicotinate(57)methylpentanoate(17)methylpropanoate(47)morpholinecarboxylate(10)naphthalene-carboxylate(1)naphthoate(41)naphthyridine-carboxylate(34)nitroisonicotinate(2)nitronicotinate(8)nitropicolinate(13)nitropyrrole-carboxylate(5)nonanedioate(4)nonanedioate(4)octanedioate(4)octanoate(15)octyl carboxylate(5)oxazole-carboxylate(105)oxoacetate(21)oxoacetate(21)oxobutanoate(56)oxobutanoate(56)oxocyclobutanecarboxylate(3)oxocyclohexanecarboxylate(11)oxocyclopentanecarboxylate(5)oxoethyl-piperidine-carboxylate(5)oxoheptanoate(8)oxoheptanoate(8)oxohexanoate(14)oxohexanoate(14)oxopentanoate(31)oxopropanoate(39)oxopropanoate(39)oxopropionate(3)oxosuccinate(11)pentaacetate(8)pentaacetate(8)pentanoate(13)pentanoate(13)pentyl carboxylate(3)phenyl-boronic-acid-carboxylate(4)phenyl-pyrazole-carboxylate(39)phenylacetate(26)phenylacrylate(5)phenylphosphonate(4)phenylpropanoate(42)phenylpropionate(3)piperazine-carboxylate(50)piperidine-carboxylate-hydrochloride(55)pivalate(9)propanoate(296)propiolate(5)propionyloxy(2)propoxycarbonyl(9)propylpentanoate(5)propylpentanoate(5)pyranyl-carboxylate(7)pyrazinecarboxylate(95)pyrazole-carboxylate(40)pyrazolopyridine-carboxylate(63)pyridazine-carboxylate(58)pyridinecarboxylate(13)pyrimidinecarboxylate(4)pyrimidinecarboxylic(3)pyrrole-carboxylate(43)pyrrolidine-carboxylate(31)quinazine-carboxylate(3)quinazoline-carboxylate(4)quinoline-carboxylate(280)quinoxaline-carboxylate(5)stearate(4)succinate(24)succinate(24)sulfamoylbenzoate(6)tert-butyl acetate(27)tert-butyl carbonate(7)tert-butyl hydrazinecarboxylate(7)tert-butyl oxyethylcarbamate(6)tert-butyl propanoate(3)tetraacetate(7)tetraacetate(7)tetradecanoate(10)tetradecanoate(10)tetrahydro-carboxylate(144)tetrahydrofuran-carboxylate(26)tetrahydroisoquinoline-carboxylate(16)tetrahydroquinoline-carboxylate(3)thiadiazole-carboxylate(14)thiazolecarboxylate(3)thiodiacetate(3)thiomorpholine-carboxylate(2)thiophenecarboxylate(226)toluenesulfonate(26)tosylcarbamate(4)tosyloxy(12)triacetate(12)triacetate(12)triacrylate(2)triazole-carboxylate(37)tricarboxylate(21)trifluoromethyl-nicotinate(4)trifluoromethyl-phenyl-acetate(15)trimethylolpropane(2)vinylbenzoate(3)vinylcyclopropanecarboxylic(5)vinylcyclopropanecarboxylic acid(3)
Ethers(82191)
(1S,4aR,7aR)-1-(((S)-tetrahydro-2H-pyran-2-yl)oxy)-1,4a,5,7a-tetrahydrocyclopenta[c]pyran(4)(2,2,2-trifluoroethoxy)benzene(12)(2-(benzyloxy)phenyl)boronic acid(23)(2-(phenoxymethyl)phenyl)boronic acid(3)(2-chloro-3-methoxyphenyl)boronic acid(4)(2-chloroethoxy)benzene(4)(2-methoxyethoxy)benzene(18)(3-(benzyloxy)phenyl)boronic acid(9)(3-(phenoxymethyl)phenyl)boronic acid(3)(3-chloro-2-methoxyphenyl)boronic acid(4)(3-phenoxyphenyl)methanol(3)(4-(benzyloxy)phenyl)boronic acid(12)(4-(phenoxymethyl)phenyl)boronic acid(6)(4-methoxyphenyl)(phenyl)methanone(7)(9H-fluoren-9-yl)methyl (2-(benzyloxy)ethyl)carbamate(6)(9H-fluoren-9-yl)methyl (4-methoxybenzyl)carbamate(7)(allyloxy)benzene(15)(ethoxymethyl)benzene(6)(hexyloxy)benzene(5)(methoxymethyl)benzene(17)(prop-2-yn-1-yloxy)benzene(8)(R)-2-phenoxytetrahydro-2H-pyran(9)(S)-7-(((S)-tetrahydro-2H-pyran-2-yl)oxy)-7,8,9,10-tetrahydrotetracene-5,12-dione(5)1,2-bis(benzyloxy)benzene(11)1,2-dichloro-3-methoxybenzene(4)1,2-dichloro-4-methoxybenzene(8)1,2-difluoro-3-methoxybenzene(23)1,2-difluoro-4-methoxybenzene(12)1,2-dimethoxybenzene(131)1,2-diphenoxyethane(11)1,3-bis(benzyloxy)benzene(8)1,3-dibromo-2-methoxybenzene(8)1,3-dichloro-2-methoxybenzene(7)1,3-diiodo-2-phenoxybenzene(3)1,3-dimethoxybenzene(140)1,3-diphenoxybenzene(8)1,4-dichloro-2-methoxybenzene(6)1,4-dimethoxybenzene(30)1-(2-methoxyphenyl)ethan-1-one(23)1-(3-(benzyloxy)phenyl)ethan-1-one(3)1-(3-methoxyphenyl)ethan-1-one(24)1-(3-methoxyphenyl)piperazine(4)1-(4-(benzyloxy)phenyl)ethan-1-one(4)1-(4-methoxybenzyl)-1H-pyrazole(5)1-(benzyloxy)-2-bromobenzene(12)1-(benzyloxy)-2-chlorobenzene(16)1-(benzyloxy)-2-fluorobenzene(10)1-(benzyloxy)-2-methoxybenzene(19)1-(benzyloxy)-2-methylbenzene(12)1-(benzyloxy)-2-nitrobenzene(3)1-(benzyloxy)-3-bromobenzene(12)1-(benzyloxy)-3-chlorobenzene(6)1-(benzyloxy)-3-fluorobenzene(14)1-(benzyloxy)-3-methylbenzene(8)1-(benzyloxy)-3-nitrobenzene(10)1-(benzyloxy)-4-bromobenzene(8)1-(benzyloxy)-4-iodobenzene(3)1-(bromomethyl)-2-methoxybenzene(12)1-(bromomethyl)-3-methoxybenzene(17)1-(difluoromethoxy)-2-fluorobenzene(9)1-benzyl-4-methoxybenzene(7)1-bromo-2,3-dimethoxybenzene(6)1-bromo-2-chloro-3-methoxybenzene(6)1-bromo-2-ethoxybenzene(16)1-bromo-2-fluoro-3-methoxybenzene(7)1-bromo-2-phenoxybenzene(3)1-bromo-3-methoxybenzene(78)1-bromo-3-phenoxybenzene(8)1-bromo-4-phenoxybenzene(8)1-chloro-2,4-dimethoxybenzene(4)1-chloro-2-ethoxybenzene(8)1-chloro-2-phenoxybenzene(10)1-chloro-2-propoxybenzene(4)1-chloro-3-fluoro-2-methoxybenzene(3)1-chloro-3-methoxybenzene(65)1-chloro-3-phenoxybenzene(5)1-chloro-4-methoxybenzene(50)1-chloro-4-phenoxybenzene(13)1-cyclopropyl-3-(4-(quinolin-4-yloxy)phenyl)urea(6)1-ethoxy-2,3-difluorobenzene(8)1-ethoxy-2-fluorobenzene(32)1-ethoxy-3-nitrobenzene(5)1-fluoro-2-(trifluoromethoxy)benzene(18)1-fluoro-2-isopropoxybenzene(11)1-fluoro-2-phenoxybenzene(11)1-fluoro-3-methoxybenzene(95)1-fluoro-3-phenoxybenzene(8)1-fluoro-4-(trifluoromethoxy)benzene(11)1-fluoro-4-methoxybenzene(53)1-fluoro-4-phenoxybenzene(8)1-iodo-2-methoxybenzene(48)1-iodo-3-methoxybenzene(23)1-iodo-4-methoxybenzene(15)1-iodo-4-phenoxybenzene(4)1-methoxy-2-(trifluoromethyl)benzene(14)1-methoxy-3-(trifluoromethoxy)benzene(5)1-methoxy-3-(trifluoromethyl)benzene(23)1-methoxy-3-nitrobenzene(59)1-methoxy-4-(phenoxymethyl)benzene(3)1-methoxy-4-(trifluoromethyl)benzene(11)1-methoxy-4-nitrobenzene(38)1-methoxynaphthalene(18)1-methyl-3-phenoxybenzene(9)1-nitro-2-phenoxybenzene(4)1-nitro-4-phenoxybenzene(5)1-phenoxy-3-(trifluoromethyl)benzene(2)1-phenoxy-4-(trifluoromethyl)benzene(2)1-phenyl-3-(4-(pyridin-4-yloxy)phenyl)urea(8)2,3,5,6,8,9,11,12-octahydrobenzo[b][1,4,7,10,13]pentaoxacyclopentadecine(8)2,3-dimethoxypyridine(7)2,4,6-triphenoxy-1,3,5-triazine(4)2,4-dibromo-1-methoxybenzene(4)2,4-dichloro-1-methoxybenzene(11)2,4-difluoro-1-methoxybenzene(14)2,4-dimethoxy-1-nitrobenzene(4)2,4-dimethoxypyrimidine(13)2,6-dimethoxypyridine(11)2-(2-methoxyphenyl)acetic acid(9)2-(3-isopropoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(5)2-(3-methoxyphenyl)acetic acid(14)2-(4-(benzyloxy)phenyl)acetonitrile(3)2-(4-methoxyphenyl)acetic acid(17)2-(benzyloxy)benzaldehyde(13)2-(benzyloxy)naphthalene(5)2-(benzyloxy)phenol(5)2-(methoxymethyl)morpholine(4)2-(methoxymethyl)pyrrolidine(6)2-(phenoxymethyl)oxirane(13)2-(phenoxymethyl)pyridine(5)2-(phenoxymethyl)tetrahydro-2H-pyran(5)2-bromo-1,3-dimethoxybenzene(6)2-bromo-1,4-dimethoxybenzene(8)2-bromo-1-fluoro-4-methoxybenzene(12)2-bromo-1-methoxy-3-methylbenzene(3)2-bromo-1-methoxy-4-methylbenzene(4)2-bromo-3-methoxypyrazine(5)2-bromo-4-methoxy-1-nitrobenzene(6)2-bromo-4-methoxyaniline(5)2-bromo-4-methoxybenzaldehyde(4)2-bromo-4-methoxybenzoic acid(4)2-bromo-5-methoxybenzaldehyde(4)2-bromo-6-methoxyaniline(4)2-bromo-6-methoxyphenol(5)2-chloro-1,3-dimethoxybenzene(3)2-chloro-1,4-dimethoxybenzene(3)2-chloro-3-methoxybenzaldehyde(4)2-chloro-3-methoxybenzoic acid(4)2-chloro-4-fluoro-1-methoxybenzene(6)2-chloro-4-methoxypyrimidine(14)2-chloro-5-methoxyaniline(5)2-chloro-6-methoxyaniline(3)2-chloro-6-methoxyphenol(4)2-ethoxynaphthalene(4)2-ethoxypyridine(20)2-ethoxypyrimidine(7)2-fluoro-1,3-dimethoxybenzene(7)2-fluoro-1-methoxy-4-nitrobenzene(6)2-Fluoro-4-methoxyaniline(5)2-fluoro-5-methoxybenzoic acid(5)2-fluoro-5-methoxypyridine(4)2-fluoro-6-methoxypyridine(6)2-isopropoxypyridine(16)2-methoxy-1,1'-biphenyl(7)2-methoxy-1,3,5-triazine(10)2-methoxy-4-methylpyridine(5)2-methoxy-5-methylpyridine(5)2-methoxy-6-methylpyridine(14)2-methoxy-6-nitrophenol(5)2-methoxybenzamide(5)2-methoxybenzenesulfonyl chloride(15)2-methoxynaphthalene(23)2-methoxyphenol(134)2-methoxypyrazine(27)2-methoxypyridine(197)2-methoxypyrimidin-4-amine(6)2-methoxypyrimidine(43)2-methoxythiophene(7)2-methyl-N-phenylaniline(7)2-phenoxyacetonitrile(8)2-phenoxyaniline(4)2-phenoxybenzonitrile(5)2-phenoxyethan-1-ol(12)2-phenoxypropanoic acid(11)2-phenoxypyrimidine(8)2-phenoxytetrahydro-2H-pyran(6)2-phenoxythiazole(5)2-propoxypyridine(9)3,5-dimethoxypyridine(4)3-(3-methoxyphenyl)propanoic acid(7)3-(benzyloxy)aniline(10)3-(benzyloxy)benzaldehyde(12)3-(benzyloxy)benzoic acid(5)3-(benzyloxy)phenol(5)3-(methoxymethyl)morpholine(4)3-chloro-2-methoxybenzoic acid(4)3-chloro-4-methoxyaniline(5)3-chloro-4-methoxybenzaldehyde(3)3-chloro-4-methoxybenzoic acid(4)3-ethoxypyridine(12)3-fluoro-2-methoxypyridine(14)3-fluoro-4-methoxybenzoic acid(5)3-methoxypiperidine(7)3-methoxypyridazine(14)3-methoxypyridine(52)3-methoxythiophene(7)3-phenoxyaniline(2)3-phenoxybenzaldehyde(4)3-phenoxybenzoic acid(6)3-phenoxybenzonitrile(4)3-phenoxypropanoic acid(6)3-phenoxypyrrolidine(5)4-(3-phenoxypropyl)morpholine(7)4-(benzyloxy)aniline(5)4-(benzyloxy)benzaldehyde(15)4-(benzyloxy)benzoic acid(5)4-(benzyloxy)phenol(3)4-(difluoromethoxy)aniline(5)4-(dimethoxymethyl)pyrimidine(5)4-(phenylethynyl)phenol(7)4-chloro-2-methoxypyrimidine(12)4-chloro-5-methoxypyrimidine(7)4-chloro-6-methoxypyrimidine(5)4-ethoxypyridine(7)4-ethoxypyrimidine(6)4-methoxy-1,1'-biphenyl(4)4-methoxy-1H-indazole(7)4-methoxy-1H-indole(11)4-methoxy-2-methylpyrimidine(8)4-methoxy-N-phenylaniline(5)4-methoxy-N-phenylbenzenesulfonamide(3)4-methoxybenzenesulfonamide(5)4-methoxypyridazine(5)4-methoxypyridine(67)4-methoxypyrimidin-2-amine(6)4-methoxypyrimidine(50)4-phenoxyaniline(6)4-phenoxybenzaldehyde(7)4-phenoxybenzoic acid(4)4-phenoxypyridine(7)4-phenoxypyrimidine(5)4-phenoxyquinoline(5)4-phenoxytetrahydro-2H-pyran(5)5-(benzyloxy)pyrimidine(5)5-fluoro-2-methoxypyridine(15)5-methoxy-1H-benzo[d]imidazole(2)5-methoxy-1H-indazole(5)5-methoxy-1H-indole(11)5-methoxy-2-methyl-1H-indole(5)5-methoxy-2-nitroaniline(5)5-methoxyindoline(4)5-methoxyisobenzofuran-1(3H)-one(5)5-methoxyisoquinoline(6)6-methoxy-1,2,3,4-tetrahydroisoquinoline(3)6-methoxy-1H-indazole(14)6-methoxy-1H-indole(12)6-methoxy-1H-pyrrolo[2,3-b]pyridine(3)6-methoxy-3,4-dihydroisoquinolin-1(2H)-one(3)6-methoxybenzo[d]thiazole(6)6-methoxybenzofuran(4)6-methoxychromane(5)6-methoxyisoindolin-1-one(3)6-methoxyquinoxaline(6)7-(benzyloxy)quinoline(6)7-methoxy-1,2,3,4-tetrahydroisoquinoline(6)7-methoxy-1H-indazole(5)7-methoxy-1H-indole(8)7-methoxy-3,4-dihydroquinolin-2(1H)-one(3)7-methoxyisoquinoline(5)8-methoxyisoquinoline(7)8-methoxyquinazoline(3)butoxybenzene(24)ethoxybenzene(139)ethyl 2-phenoxyacetate(8)isobutoxybenzene(12)isopropoxybenzene(51)methoxycyclohexane(5)methyl 2-methoxybenzoate(44)methyl 2-phenoxyacetate(9)methyl 2-phenoxybenzoate(4)methyl 3-(benzyloxy)benzoate(5)methyl 3-methoxybenzoate(36)methyl 4-phenoxybenzoate(3)N,N-dimethyl-2-phenoxyethan-1-amine(12)N-(2-phenoxyphenyl)methanesulfonamide(4)N-(4-phenoxyphenyl)acetamide(4)N-phenyl-N-(4-(quinolin-4-yloxy)phenyl)cyclopropane-1,1-dicarboxamide(5)phenethoxybenzene(23)propoxybenzene(33)tert-butoxybenzene(15)tert-butyl 3-methoxypyrrolidine-1-carboxylate(6)tert-butyl 4-phenoxypiperidine-1-carboxylate(12)tert-butyl 5-methoxy-1H-indole-1-carboxylate(6)(2-ethylhexyl)oxy(8)(S)-2-methoxyethanamine(6)(trimethylsilyl)ethoxy(13)2,2,2-trifluoroethoxy(44)2,2-difluoroethoxy(4)2,2-dimethoxyethyl(6)2-bromo-1,1,2,2-tetrafluoroethoxy(5)2-bromoethoxy(14)2-methoxyethoxy(52)2-methoxyethoxymethyl(6)2-methoxyethyl(25)2-methyl-2-oxypropanoic acid(8)2-oxoacetic acid(26)2-oxy-propanoic acid(9)3-(oxy)propanoic acid(13)3-methoxypropoxy(17)4-bromobutoxy(2)allyloxy(28)benzo-crown-ether(5)butoxy(64)butoxy-phenyl-boronic acid(69)butoxy-phenyl-boronic acid/ester(14)butoxy-phenyl-boronic-acid(1)chloroanisole(3)cyanomethoxy(10)decyloxy(8)diethoxy(14)diethoxy(14)diethoxyethyl(9)diethoxyethyl(9)diethoxymethyl(8)diethoxymethyl(8)diethylaminoethoxy(4)dimethoxyethyl(8)dimethoxymethyl(20)dimethoxypropane(11)dimethoxypropane(11)dimethylamino ethoxy(25)dimethylaminopropoxy(5)dioxane(153)dioxepine(7)dioxepine(7)dioxopropan(1)dioxopropan(1)disulfide(38)dodecyloxy(11)ethoxyethanol(29)ethyl 2-oxyacetate(16)fluoromethoxy-nitro-benzene(50)heptyloxy(9)hexyloxy(16)isobutoxy(31)isopentyloxy(4)isopropoxy(153)isopropoxyphenylboronic(4)methoxy-acetic acid(5)methoxy-carbazole(8)methoxy-chromen-one(6)methoxy-indanone(37)methoxy-indazole-carboxylate(1)methoxy-inden-amine(13)methoxy-indole-carboxylate(27)methoxy-methyl-indole(87)methoxy-methylbenzene(90)methoxy-naphthyridine(6)methoxy-pyran(144)methoxy-pyrazolo-pyridine(30)methoxy-trifluoromethyl-benzene(22)methoxyacetate(4)methoxychroman(17)methoxyethoxyethoxyethoxy(3)methoxyphenylboronic-acid(12)methyl 2-oxy-acetate(14)nitroanisole(13)octyloxy(17)oxy ethanamine(15)oxy-acetohydrazide(5)oxyacetic acid(81)oxypropane-1,2-dio(7)prop-2-yn-1-oxy(25)propoxy(80)tert-butoxy(42)tert-butoxybromobenzene(6)tert-butoxymethyl(6)tetrahydroquinoline-methoxy(5)
Fluorinated Building Blocks(96211)
(2,3-difluorophenyl)boronic acid(16)(2,6-difluorophenyl)boronic acid(13)(2-chloro-6-fluorophenyl)boronic acid(6)(2-fluorobenzyl)hydrazine(5)(2-fluorophenyl)(phenyl)methanone(11)(2-fluorophenyl)boronic acid(72)(2-fluorophenyl)hydrazine(13)(2-fluorophenyl)methanamine(30)(2-fluorophenyl)methanol(47)(3-fluorophenyl)(phenyl)methanone(5)(3-fluorophenyl)boronic acid(96)(3-fluorophenyl)methanol(39)(4-fluorophenyl)(phenyl)methanone(8)(4-fluorophenyl)boronic acid(43)(4-fluorophenyl)methanamine(17)(4-fluorophenyl)methanol(18)1,1-difluorocyclobutane(14)1,1-difluorocyclohexane(16)1,2,3,4,5-pentafluorobenzene(44)1,2,3,4-tetrafluorobenzene(52)1,2,3-trifluorobenzene(104)1,2,4-trifluorobenzene(114)1,2-dibromo-3-fluorobenzene(7)1,2-dibromo-4-fluorobenzene(9)1,2-dichloro-3-fluorobenzene(8)1,2-dichloro-4-fluorobenzene(8)1,2-difluoro-3-iodobenzene(10)1,2-difluoro-3-methoxybenzene(23)1,2-difluoro-3-methylbenzene(17)1,2-difluoro-3-nitrobenzene(15)1,2-difluoro-4-methoxybenzene(12)1,2-difluoro-4-nitrobenzene(20)1,2-difluorobenzene(288)1,3-dibromo-2,4-difluorobenzene(5)1,3-dibromo-2-fluorobenzene(11)1,3-dichloro-2-fluorobenzene(17)1,3-difluoro-2-iodobenzene(12)1,3-difluoro-2-methylbenzene(10)1,3-difluoro-2-nitrobenzene(10)1,4-dichloro-2-fluorobenzene(8)1,4-difluoro-2-iodobenzene(7)1,4-difluoro-2-nitrobenzene(17)1,4-difluorobenzene(174)1-(2-fluorophenyl)ethan-1-amine(40)1-(2-fluorophenyl)ethan-1-one(34)1-(2-fluorophenyl)piperazine(8)1-(2-fluorophenyl)propan-1-amine(5)1-(3-fluorophenyl)ethan-1-one(41)1-(4-fluorophenyl)ethan-1-one(19)1-(benzyloxy)-2-fluorobenzene(10)1-(benzyloxy)-3-fluorobenzene(14)1-(bromomethyl)-2-fluorobenzene(39)1-(bromomethyl)-3-fluorobenzene(32)1-(chloromethyl)-2-fluorobenzene(5)1-(chloromethyl)-3-fluorobenzene(8)1-benzyl-4-fluorobenzene(2)1-bromo-2,3-difluorobenzene(53)1-bromo-2,4-difluorobenzene(37)1-bromo-2-chloro-3-fluorobenzene(14)1-bromo-2-fluoro-3-methylbenzene(10)1-bromo-3-fluoro-2-methoxybenzene(12)1-bromo-3-fluoro-2-methylbenzene(16)1-bromo-3-fluoro-2-nitrobenzene(10)1-bromo-4-fluoro-2-methoxybenzene(8)1-bromo-4-fluoro-2-methylbenzene(12)1-chloro-2,3-difluorobenzene(25)1-chloro-2,4-difluorobenzene(19)1-chloro-3-fluoro-2-methoxybenzene(3)1-chloro-3-fluoro-2-methylbenzene(5)1-cyclohexyl-2-fluorobenzene(4)1-ethoxy-2,3-difluorobenzene(8)1-ethoxy-2-fluorobenzene(32)1-ethynyl-2-fluorobenzene(6)1-fluoro-2,3-dimethylbenzene(7)1-fluoro-2-(methylsulfonyl)benzene(3)1-fluoro-2-iodo-4-methylbenzene(3)1-fluoro-2-iodobenzene(70)1-fluoro-2-isopropoxybenzene(11)1-fluoro-2-methylbenzene(109)1-fluoro-2-phenoxybenzene(11)1-fluoro-3,5-dimethylbenzene(10)1-fluoro-3-(methylsulfonyl)benzene(9)1-fluoro-3-iodo-2-methylbenzene(3)1-fluoro-3-iodobenzene(62)1-fluoro-3-methoxybenzene(95)1-fluoro-3-methylbenzene(120)1-fluoro-3-nitrobenzene(88)1-fluoro-3-phenoxybenzene(8)1-fluoro-4-iodobenzene(34)1-fluoro-4-methoxybenzene(53)1-fluoro-4-methylbenzene(66)1-fluoro-4-phenoxybenzene(8)1-fluorobicyclo[1.1.1]pentane(4)1-fluoronaphthalene(23)2'-fluoro-1,1':4',1''-terphenyl(6)2,2',3,3',5,5',6,6'-octafluoro-1,1'-biphenyl(4)2,3-difluoroaniline(25)2,3-difluorobenzaldehyde(9)2,3-difluorobenzoic acid(16)2,3-difluorobenzonitrile(17)2,3-difluorophenol(33)2,3-difluoropyridine(13)2,4-dibromo-1-fluorobenzene(9)2,4-dichloro-1-fluorobenzene(18)2,4-difluoro-1-iodobenzene(9)2,4-difluoro-1-methoxybenzene(14)2,4-difluoro-1-nitrobenzene(29)2,4-difluoroaniline(25)2,4-difluorobenzaldehyde(14)2,4-difluorobenzoic acid(15)2,4-difluorobenzonitrile(10)2,4-difluorophenol(18)2,5-difluoroaniline(23)2,5-difluorobenzoic acid(17)2,5-difluoropyridine(8)2,6-difluoroaniline(15)2,6-difluorobenzaldehyde(16)2,6-difluorobenzoic acid(25)2,6-difluorobenzonitrile(21)2,6-difluorophenol(18)2,6-difluoropyridine(29)2-(2-fluorophenyl)acetic acid(34)2-(2-fluorophenyl)acetonitrile(15)2-(2-fluorophenyl)pyrrolidine(16)2-(3-fluorophenyl)acetic acid(28)2-(3-fluorophenyl)pyrrolidine(5)2-(4-fluorophenyl)-1H-indole(4)2-(4-fluorophenyl)acetic acid(17)2-(bromomethyl)-1,3-difluorobenzene(11)2-amino-2-(2-fluorophenyl)acetic acid(11)2-amino-3-(2-fluorophenyl)propanoic acid(15)2-amino-3-fluorobenzoic acid(6)2-bromo-1,3,4-trifluorobenzene(16)2-bromo-1,3-difluorobenzene(26)2-bromo-1,4-difluorobenzene(36)2-bromo-1-chloro-3-fluorobenzene(8)2-bromo-1-chloro-4-fluorobenzene(13)2-bromo-1-fluoro-4-methylbenzene(11)2-bromo-4-chloro-1-fluorobenzene(10)2-bromo-4-fluoro-1-methoxybenzene(9)2-bromo-4-fluoro-1-methylbenzene(16)2-bromo-4-fluoroaniline(19)2-bromo-4-fluorobenzaldehyde(5)2-bromo-4-fluorobenzonitrile(4)2-bromo-4-fluorophenol(7)2-bromo-5-fluoroaniline(13)2-bromo-5-fluorobenzaldehyde(4)2-bromo-5-fluorobenzoic acid(3)2-bromo-6-fluoroaniline(13)2-bromo-6-fluorophenol(8)2-chloro-1,3-difluorobenzene(22)2-chloro-1,4-difluorobenzene(17)2-chloro-3-fluoropyridine(20)2-chloro-4-fluoro-1-methoxybenzene(6)2-chloro-4-fluoroaniline(10)2-chloro-4-fluorobenzaldehyde(8)2-chloro-4-fluorobenzoic acid(5)2-chloro-4-fluorobenzonitrile(6)2-chloro-4-fluorophenol(9)2-chloro-5-fluoropyrimidine(6)2-chloro-6-fluoroaniline(6)2-chloro-6-fluorobenzoic acid(6)2-chloro-6-fluorobenzonitrile(4)2-chloro-6-fluorophenol(4)2-fluoro-1,3-dimethoxybenzene(7)2-fluoro-1,3-dimethylbenzene(12)2-fluoro-1-iodo-4-methylbenzene(4)2-fluoro-3-methylbenzaldehyde(7)2-fluoro-3-methylpyridine(14)2-Fluoro-4-methoxyaniline(5)2-fluoro-4-methyl-1-nitrobenzene(4)2-fluoro-4-methylbenzoic acid(6)2-fluoro-4-methylpyridine(6)2-fluoro-5-methylpyridine(6)2-fluoro-6-methylpyridine(20)2-fluoro-N-methylbenzamide(8)2-fluoro-N-phenylaniline(4)2-fluoro-N-phenylbenzenesulfonamide(5)2-fluoroaniline(113)2-fluorobenzaldehyde(75)2-fluorobenzamide(22)2-fluorobenzenesulfonamide(8)2-fluorobenzenesulfonyl chloride(31)2-fluorobenzoic acid(71)2-fluoropyridine(15)2-fluoropyrimidine(9)3,3-difluoropiperidine(29)3,3-difluoropyrrolidine(27)3,4-difluoroaniline(26)3,4-difluorophenol(13)3,5-difluoropyridine(12)3-((tert-butoxycarbonyl)amino)-4-(2-fluorophenyl)butanoic acid(8)3-chloro-2-fluoroaniline(8)3-fluoro-1,1'-biphenyl(16)3-fluoro-2-iodoaniline(8)3-fluoro-2-methylaniline(8)3-fluoro-2-methylpyridine(15)3-fluoro-4-nitroaniline(6)3-fluoroaniline(129)3-fluorobenzaldehyde(58)3-fluorobenzamide(15)3-fluorobenzenesulfonyl chloride(22)3-fluorobenzoic acid(103)3-fluorophenol(92)3-fluoropiperidine(26)3-fluoropyridin-2-amine(12)3-fluoropyridin-4-amine(4)3-fluorothiophene(6)4,4-difluoropiperidine(5)4-(2-fluorophenyl)morpholine(8)4-(4-fluorophenyl)thiazole(9)4-chloro-5-fluoro-2-methylpyrimidine(5)4-fluoro-1-methyl-2-nitrobenzene(7)4-fluoro-1H-indazole(22)4-fluoro-1H-indole(24)4-fluoro-1H-pyrrolo[2,3-b]pyridine(6)4-fluoro-2-iodoaniline(6)4-fluoro-2-nitroaniline(9)4-fluoroaniline(121)4-fluorobenzaldehyde(44)4-fluorobenzamide(10)4-fluorobenzenesulfonyl chloride(25)4-fluorobenzo[d]thiazole(6)4-fluorobenzoic acid(56)4-fluorobenzonitrile(43)4-fluoroisoquinoline(6)4-fluorophenol(63)4-fluoropyrimidine(8)5-fluoro-1,2,3,4-tetrahydroisoquinoline(3)5-fluoro-1H-benzo[d][1,2,3]triazole(4)5-fluoro-1H-benzo[d]imidazole(7)5-fluoro-1H-indole(21)5-fluoro-1H-pyrrolo[2,3-b]pyridine(9)5-fluoro-2,3-dihydro-1H-indene(3)5-fluoro-2,3-dihydrobenzofuran(5)5-fluoro-2-methylpyridine(8)5-fluoro-3,4-dihydronaphthalen-1(2H)-one(5)5-fluorochromane(6)5-fluoroindoline(3)5-fluoroisoindolin-1-one(3)5-fluoropyridin-2-amine(15)5-fluoropyrimidine(11)6-fluoro-1,2,3,4-tetrahydroisoquinoline(3)6-fluoro-1,2,3,4-tetrahydroquinoline(3)6-fluoro-1H-indazole(15)6-fluoro-1H-indole(17)6-fluoro-1H-pyrrolo[2,3-b]pyridine(3)6-fluorobenzo[d]thiazole(5)6-fluorochromane(6)6-fluoropyridin-2-amine(8)6-fluoropyridin-3-amine(4)7-fluoro-1,2,3,4-tetrahydroisoquinoline(3)7-fluoro-1,2,3,4-tetrahydroquinoline(3)7-fluoro-1H-indazole(12)7-fluoro-1H-indole(18)7-fluorobenzo[d]thiazole(9)7-fluoroindoline-2,3-dione(6)7-fluoroisoquinoline(3)8-fluoro-3,4-dihydronaphthalen-1(2H)-one(5)8-fluorochromane(5)8-fluoroimidazo[1,2-a]pyridine(3)8-fluoroisoquinoline(10)8-fluoroquinazoline(4)benzenesulfonyl fluoride(51)ethyl 2-fluorobenzoate(26)ethyl 3-fluorobenzoate(17)fluorobenzene(4368)methyl 2,6-difluorobenzoate(13)methyl 2-fluorobenzoate(46)methyl 3-fluorobenzoate(80)methyl 4-fluorobenzoate(44)methyl 6-fluoronicotinate(4)N-(3-fluorophenyl)acetamide(6)N-(4-fluorophenyl)benzamide(3)pentafluoro(phenyl)-l6-sulfane(19)phenyl trifluoromethanesulfonate(20)tert-butyl (4-fluorophenyl)carbamate(5)trifluoro(phenyl)borate(39)1,1-difluoroethyl(10)2,2-difluoroacetic acid(9)2,2-difluoroethoxy(4)2,2-difluoroethyl(9)2-bromo-1,1,2,2-tetrafluoroethoxy(5)2-fluoroethyl(2)bromo-fluorobenzene(287)carbonyl fluoride(8)chloro-fluoro-benzene(69)dichlorofluorobenzene(23)difluoro(901)difluoroaniline(37)difluorobenzaldehyde(33)difluorobenzamide(6)difluorobenzenesulfonamide(5)difluorobenzoate(59)difluorobenzylamine(1)difluorocyclobutanecarboxylate(4)difluoroethyl(27)difluoroethyl(27)difluoromethoxy(200)difluoromethoxy(200)difluoropiperidine(19)difluoropropan(7)difluoropropan(7)difluoropyrimidine(6)difluoroquinoline(9)ethyl 2,2-difluoro-acetate(8)fluindamine(12)fluindenamine(38)fluoro-benzenesulfonyl-chloride(32)fluoro-indazol-amine(24)fluoro-indazole-carboxylate(20)fluoro-indole-carboxylate(54)fluoro-methyl(7)fluoro-methyl-indazole(74)fluoro-naphthyridine(2)fluoro-phenyl-boronic acid(509)fluoro-pyrrolopyridine(64)fluoroacetanilide(5)fluoroacetophenone(8)fluoroaniline(65)fluoroanisole(9)fluorobenzaldehyde(69)fluorobenzamide(28)fluorobenzenesulfonamide(7)fluorobenzenethiol(6)fluorobenzimidamide(5)fluorobenzo(71)fluorobenzoate(85)fluorobenzoic(82)fluorobenzoyl(38)fluorobenzoyl-chloride(22)fluorobenzylamine(10)fluorobromobenzene(3)fluorocarboxylic acid(6)fluorochroman(27)fluorocinnamic(10)fluorocinnamic(10)fluorocinnamic acid(2)fluorocyclopropanecarboxylic(9)fluoroethoxy(4)fluoroethyl(8)fluoroethyl(8)fluoroimidazo(24)fluoroindan(3)fluoroindan(3)fluoroisonicotinaldehyde(7)fluoroisonicotinate(8)fluoroisonicotinic(14)fluoroisonicotinic acid(7)fluoromethyl(15)fluoromethyl(15)fluoromethyl-indole(102)fluoronicotinaldehyde(20)fluoronicotinamide(3)fluoronicotinate(19)fluoronicotinic(19)fluoronicotinic(19)fluoronicotinic acid(7)fluoronitrobenzene(14)fluorophenyl(2)fluorophenyl-boronate(141)fluorophenylisocyanate(3)fluorophenylisothiocyanate(2)fluoropropan(15)fluoropropan(15)fluoropyrimidine(32)fluoroquinazoline(20)fluoroquinoline(112)fluorostyryl(7)fluorosulfonyl(12)fluorosulfonyl(12)fluorotoluene(7)heptadecafluorodecyl(2)heptafluoro(10)hexafluoro(9)hexafluoro(9)hexafluoropropan(4)hexafluoropropan(4)nonafluoro(2)nonafluoro(2)nonafluorobutane(1)nonafluorobutane(1)nonafluorohexyl(3)nonafluorohexyl(3)octafluoro(2)octafluoro(2)pentafluoro(4)pentafluorobenzoate(4)perfluorohexyl(1)perfluorohexyl(1)tetrafluoro(12)tetrafluoro(12)tetrafluoroaniline(7)tetrafluoroethoxy(3)tetrafluoroethoxy(3)tetrafluoropropyl(3)tetrafluoropropyl(3)tetrahydroquinoline-fluoro(11)tridecafluorooctyl(1)tridecafluorooctyl(1)trifluoro(271)trifluoro(271)trifluoroacetamide(8)trifluoroacetyl(13)trifluoroaniline(11)trifluorobenzaldehyde(9)trifluorobenzoate(14)trifluorobenzoylchloride(2)trifluorobut(29)trifluorobut(29)trifluorobutane(19)trifluorobutane(19)trifluoroethan(2)trifluoroethan(2)trifluoroethanamine(23)trifluoroethanol(6)trifluoroethoxy(60)trifluoroethoxy(60)trifluoroethyl(63)trifluoromethanesulfonamide(4)trifluoromethanesulfonyl(22)trifluoromethoxy(480)trifluoromethoxy(480)trifluoromethylthio(5)trifluoromethylthio(5)trifluoropropane(36)trifluoropropanoate(5)trifluoropropyl(9)
Hydrazines(6264)
(2-bromophenyl)hydrazine(7)(2-chlorophenyl)hydrazine(13)(2-fluorobenzyl)hydrazine(5)(2-fluorophenyl)hydrazine(13)(3-chlorophenyl)hydrazine(7)2-hydrazineylpyridine(32)2-hydrazineylpyrimidine(6)3-hydrazineylpyridine(8)4-hydrazineylpiperidine(5)isonicotinohydrazide(7)phenylhydrazine(97)picolinohydrazide(5)dichlorophenylhydrazine(3)hydrazinecarbothioamide(2)hydrazinecarboxamide(1)hydrazinecarboxylate(12)hydrazinylbenzenesulfonamide(4)hydrazinylbenzoate(6)hydrazinylmethyl(45)hydrazinylpyrimidine(3)phenyl-hydrazine-hydrochloride(83)tert-butyl hydrazinecarboxylate(7)trifluoromethyl-phenyl-hydrazine(4)
Iodides(14053)
(2-iodophenyl)(phenyl)methanone(4)(2-iodophenyl)methanol(10)(3-iodophenyl)boronic acid(4)(3-iodophenyl)methanol(6)1,2-benziodoxol-3-(1H)-one(6)1,2-difluoro-3-iodobenzene(10)1,2-diiodobenzene(10)1,3-dibromo-2-iodobenzene(4)1,3-dichloro-2-iodobenzene(5)1,3-difluoro-2-iodobenzene(12)1,3-diiodo-2-phenoxybenzene(3)1,3-diiodobenzene(41)1,4-dibromo-2-iodobenzene(3)1,4-difluoro-2-iodobenzene(7)1-(2-iodophenyl)ethan-1-one(4)1-(3-iodophenyl)ethan-1-one(3)1-(benzyloxy)-4-iodobenzene(3)1-(bromomethyl)-2-iodobenzene(9)1-benzyl-4-iodo-1H-pyrazole(5)1-bromo-2-iodobenzene(57)1-bromo-3-fluoro-2-iodobenzene(4)1-bromo-3-iodobenzene(40)1-bromo-4-iodobenzene(26)1-chloro-2-fluoro-3-iodobenzene(5)1-chloro-2-iodobenzene(51)1-chloro-3-iodo-2-methylbenzene(3)1-chloro-3-iodobenzene(46)1-chloro-4-iodobenzene(18)1-ethyl-2-iodobenzene(5)1-fluoro-2-iodo-4-methylbenzene(3)1-fluoro-2-iodobenzene(70)1-fluoro-3-iodo-2-methylbenzene(3)1-fluoro-3-iodobenzene(62)1-fluoro-4-iodobenzene(34)1-iodo-2,3-dimethylbenzene(6)1-iodo-2,4-dimethylbenzene(6)1-iodo-2-(trifluoromethoxy)benzene(6)1-iodo-2-(trifluoromethyl)benzene(12)1-iodo-2-isopropylbenzene(4)1-iodo-2-methoxybenzene(48)1-iodo-2-methylbenzene(53)1-iodo-2-nitrobenzene(32)1-iodo-3-(trifluoromethoxy)benzene(3)1-iodo-3-(trifluoromethyl)benzene(17)1-iodo-3-methoxybenzene(23)1-iodo-3-methylbenzene(32)1-iodo-3-nitrobenzene(24)1-iodo-4-methoxybenzene(15)1-iodo-4-phenoxybenzene(4)2,4-difluoro-1-iodobenzene(9)2,6-dichloro-4-iodopyridine(4)2,6-diiodoaniline(8)2,6-diiodophenol(16)2-bromo-3-iodopyridine(6)2-bromo-4-chloro-1-iodobenzene(5)2-bromo-6-iodoaniline(4)2-bromo-6-iodopyridine(5)2-chloro-3-iodopyridin-4-amine(3)2-chloro-3-iodopyridine(13)2-chloro-4-iodopyridine(19)2-chloro-5-iodopyridine(18)2-chloro-6-iodophenol(3)2-chloro-6-iodopyridine(9)2-fluoro-1-iodo-4-methylbenzene(4)2-fluoro-3-iodopyridine(8)2-fluoro-6-iodoaniline(7)2-hydroxy-3-iodobenzaldehyde(3)2-iodo-1,3-dimethylbenzene(14)2-iodo-1,4-dimethylbenzene(4)2-iodo-1-methyl-4-nitrobenzene(3)2-iodo-4-methylaniline(4)2-iodo-6-methylpyridine(5)2-iodo-6-nitroaniline(3)2-iodo-9H-fluorene(8)2-iodo-N,N-dimethylaniline(3)2-iodoaniline(52)2-iodobenzaldehyde(16)2-iodobenzamide(4)2-iodobenzene-1,3-diol(3)2-iodobenzoic acid(40)2-iodobenzonitrile(22)2-iodonaphthalene(5)2-iodophenol(52)2-iodopyrazine(25)2-iodopyridine(27)2-iodopyrimidine(8)2-iodothiophene(14)3-chloro-2-iodoaniline(3)3-chloro-4-iodoaniline(3)3-fluoro-2-iodoaniline(8)3-iodo-1-methyl-1H-pyrazole(6)3-iodo-1H-pyrrolo[2,3-b]pyridine(11)3-iodo-1H-pyrrolo[3,2-b]pyridine(6)3-iodo-2-methylaniline(3)3-iodoaniline(22)3-iodobenzaldehyde(17)3-iodobenzoic acid(22)3-iodobenzonitrile(15)3-iodoimidazo[1,2-a]pyridine(10)3-iodophenol(13)3-iodopyridazine(5)3-iodopyridin-2-amine(12)3-iodopyridin-2-ol(4)3-iodopyridin-4-amine(6)3-iodopyridin-4-ol(4)3-iodopyridine(37)3-iodotetrahydrofuran(3)4-bromo-2-chloro-1-iodobenzene(4)4-bromo-2-fluoro-1-iodobenzene(4)4-bromo-2-iodoaniline(8)4-chloro-2-iodoaniline(7)4-chloro-3-iodoaniline(3)4-chloro-5-iodopyrimidine(6)4-fluoro-2-iodoaniline(6)4-fluoro-3-iodobenzoic acid(5)4-iodo-1,1'-biphenyl(9)4-iodo-1H-pyrazole(9)4-iodo-N,N-diphenylaniline(3)4-iodopyridine(17)4-iodopyrimidine(9)5-bromo-2-iodopyridine(5)5-iodo-1H-imidazole(4)5-iodo-1H-pyrazole(4)5-iodo-7H-pyrrolo[2,3-d]pyrimidine(4)5-iodopyridin-2-amine(17)5-iodopyridin-2-ol(7)5-iodopyrimidine(5)6-chloro-5-iodopyridin-2-amine(3)ethyl 2-iodobenzoate(4)ethyl 3-iodobenzoate(8)methyl 2-iodobenzoate(24)methyl 3-iodo-4-methylbenzoate(6)methyl 3-iodobenzoate(21)methyl 4-amino-3-iodobenzoate(5)N-(2-iodophenyl)acetamide(9)phenyl(o-tolyl)iodonium(14)tert-butyl 3-iodo-1H-indole-1-carboxylate(7)3-iodopropyl(4)diiodo(19)diiodobenzoate(5)fluoro-iodo-nitro-benzene(13)fluoroiodobenzene(56)iodo-benzofuran(16)iodo-fluorene(25)iodo-indazol-amine(3)iodo-indazole(65)iodo-indazole-carboxylate(4)iodo-indole(46)iodo-isoquinoline(12)iodo-methyl-pyrazole(36)iodo-pyrazole-carboxylate(19)iodo-pyrazolopyridine(32)iodo-pyrrolopyridine(39)iodo-pyrrolopyridine-carboxylate(3)iodo-pyrrolopyrimidine(17)iodo-trifluoromethyl-pyridine(24)iodoacetate(10)iodoaniline(60)iodoanisole(5)iodobenzaldehyde(38)iodobenzamide(6)iodobenzene(170)iodobenzoate(50)iodobenzotrifluoride(6)iodobutane(4)iodobutane(4)iodoethane(3)iodoethane(3)iodoethoxy(6)iodoethoxy(6)iodoethyl(5)iodoethyl(5)iodohexane(1)iodohexane(1)iodoimidazo(19)iodomethyl(26)iodomethyl(26)iodomethyl-imidazole(9)iodomethyl-indazole(24)iodonicotinaldehyde(6)iodonicotinate(13)iodonicotinic(14)iodonicotinic(14)iodonitrobenzene(35)iodopropane(11)iodopropane(11)iodopropyl(4)iodopropyl(4)iodopyrazolo(4)iodopyrimidine(28)iodotoluene(7)
Ketones(38847)
(1E,4E)-1,5-diphenylpenta-1,4-dien-3-one(7)(1E,6E)-1,7-diphenylhepta-1,6-diene-3,5-dione(7)(2-aminophenyl)(phenyl)methanone(22)(2-bromophenyl)(phenyl)methanone(3)(2-chlorophenyl)(phenyl)methanone(17)(2-fluorophenyl)(phenyl)methanone(11)(2-hydroxyphenyl)(phenyl)methanone(22)(2-iodophenyl)(phenyl)methanone(4)(3-chlorophenyl)(phenyl)methanone(10)(3-fluorophenyl)(phenyl)methanone(5)(3-nitrophenyl)(phenyl)methanone(5)(4-aminophenyl)(phenyl)methanone(2)(4-bromophenyl)(phenyl)methanone(9)(4-chlorophenyl)(phenyl)methanone(11)(4-fluorophenyl)(phenyl)methanone(8)(4-hydroxyphenyl)(phenyl)methanone(3)(4-methoxyphenyl)(phenyl)methanone(7)(4-nitrophenyl)(phenyl)methanone(3)(R)-5-thia-1-azabicyclo[4.2.0]oct-2-en-8-one(8)1,3-dioxoisoindolin-2-yl 2-phenylacetate(5)1,3-diphenylpropan-1-one(9)1,4-diazepan-5-one(8)1,4-dihydroisoquinolin-3(2H)-one(5)1-(1H-indazol-1-yl)ethan-1-one(7)1-(1H-indol-1-yl)ethan-1-one(9)1-(1H-indol-3-yl)ethan-1-one(6)1-(1H-pyrrolo[2,3-b]pyridin-3-yl)ethan-1-one(4)1-(2-(trifluoromethyl)phenyl)ethan-1-one(7)1-(2-aminophenyl)ethan-1-one(18)1-(2-bromophenyl)ethan-1-one(23)1-(2-chlorophenyl)ethan-1-one(35)1-(2-fluorophenyl)ethan-1-one(34)1-(2-hydroxyphenyl)-2-phenylethan-1-one(5)1-(2-hydroxyphenyl)-3-phenylprop-2-en-1-one(10)1-(2-hydroxyphenyl)ethan-1-one(53)1-(2-iodophenyl)ethan-1-one(4)1-(2-methoxyphenyl)ethan-1-one(23)1-(2-nitrophenyl)ethan-1-one(13)1-(3-(benzyloxy)phenyl)ethan-1-one(3)1-(3-aminophenyl)ethan-1-one(11)1-(3-chlorophenyl)ethan-1-one(20)1-(3-fluorophenyl)ethan-1-one(41)1-(3-hydroxyphenyl)ethan-1-one(14)1-(3-iodophenyl)ethan-1-one(3)1-(3-methoxy-2-nitrophenyl)ethan-1-one(4)1-(3-methoxyphenyl)ethan-1-one(24)1-(3-nitrophenyl)ethan-1-one(13)1-(3-nitrophenyl)propan-1-one(4)1-(4-(benzyloxy)phenyl)ethan-1-one(4)1-(4-fluorophenyl)ethan-1-one(19)1-([1,1'-biphenyl]-4-yl)ethan-1-one(5)1-(benzofuran-2-yl)ethan-1-one(3)1-(imidazo[1,2-a]pyridin-3-yl)ethan-1-one(3)1-(m-tolyl)ethan-1-one(21)1-(naphthalen-2-yl)ethan-1-one(3)1-(o-tolyl)ethan-1-one(13)1-(piperidin-1-yl)ethan-1-one(13)1-(pyrazin-2-yl)ethan-1-one(5)1-(pyridin-2-yl)ethan-1-one(36)1-(pyridin-3-yl)ethan-1-one(29)1-(pyridin-4-yl)ethan-1-one(14)1-(pyrimidin-2-yl)ethan-1-one(5)1-(pyrimidin-4-yl)ethan-1-one(6)1-(pyrrolidin-1-yl)ethan-1-one(9)1-(thiazol-2-yl)ethan-1-one(9)1-(thiophen-2-yl)ethan-1-one(17)1-aminoanthracene-9,10-dione(7)1-benzylindoline-2,3-dione(5)1-benzylpiperidin-3-one(5)1-chloroanthracene-9,10-dione(6)1-hydroxyanthracene-9,10-dione(12)1-methylpiperazin-2-one(6)1-methylpyrrolidin-2-one(13)1-phenylbutan-1-one(9)1-phenylpentan-1-one(6)1-phenylpiperidin-2-one(8)1-phenylpropan-2-one(23)2,2,2-trifluoro-1-phenylethan-1-one(31)2,2-dimethyl-1-phenylpropan-1-one(6)2,2-dimethyl-2H-benzo[b][1,4]oxazin-3(4H)-one(3)2,3,4,9-tetrahydro-1H-carbazol-1-one(7)2,3-dihydrofuran-3-one(9)2,3-dihydropyridin-2-one(5)2,5-dichlorobenzoic acid(10)2-amino-1-(pyrrolidin-1-yl)ethan-1-one(5)2-Benzalacetophenone(30)2-benzoylbenzoic acid(9)2-chloro-1-phenylethan-1-one(15)2-chlorocyclohexa-2,5-diene-1,4-dione(4)2-chloroquinazolin-4(3H)-one(4)2-hydroxy-1,2-diphenylethan-1-one(5)2-hydroxy-1-phenylethan-1-one(5)2-hydroxycyclohexa-2,5-diene-1,4-dione(3)2-hydroxyquinolin-4(1H)-one(3)2-methoxycyclohexa-2,5-diene-1,4-dione(7)2-methyl-1-phenylpropan-1-one(16)2-methyl-2H-benzo[b][1,4]oxazin-3(4H)-one(3)2-methyl-3,4-dihydroisoquinolin-1(2H)-one(6)2-methylcyclohexa-2,5-diene-1,4-dione(5)2-methylcyclohexan-1-one(2)2-methylisoindolin-1-one(7)2-methylisoindoline-1,3-dione(5)2-methylpyridazin-3(2H)-one(7)2-oxo-2-phenylacetaldehyde(7)2-oxo-2-phenylacetic acid(9)3,3-difluoropiperidin-4-one(3)3,3-dimethylcyclohexan-1-one(4)3,3-dimethylpiperidin-4-one(3)3,4-dihydroquinoxalin-2(1H)-one(6)3-acetyl-2H-chromen-2-one(3)3-aminopyrrolidin-2-one(5)3-bromo-1H-pyrrole-2,5-dione(3)3-chloro-1-phenylpropan-1-one(7)3-fluoropiperidin-4-one(3)3-hydroxy-2-phenyl-4H-chromen-4-one(20)3-hydroxypyrrolidin-2-one(4)3-methylcyclohexan-1-one(7)3-methylene-2,3-dihydro-1H-inden-1-one(5)3-methylpiperazin-2-one(7)3-methylpyrimidin-4(3H)-one(6)3-oxo-3-phenylpropanenitrile(11)3-oxo-N-phenylbutanamide(8)3-phenyl-1-(pyrrolidin-1-yl)propan-1-one(5)3-phenyl-2-thioxoimidazolidin-4-one(6)3-phenylacrylaldehyde(22)3-phenylimidazolidine-2,4-dione(4)4,4,4-trifluoro-1-phenylbutane-1,3-dione(10)4-(1,2,2-triphenylvinyl)benzaldehyde(5)4-(hydroxymethyl)pyrrolidin-2-one(3)4-benzylmorpholin-3-one(6)4-bromoisoindolin-1-one(3)4-bromopyridazin-3(2H)-one(3)4-chloro-1-phenylbutan-1-one(7)4-chloropyridazin-3(2H)-one(4)4-fluoroindoline-2,3-dione(3)4-hydroxy-2H-chromen-2-one(10)4-hydroxydihydrofuran-2(3H)-one(14)4-hydroxypiperidin-2-one(3)4-hydroxypyrrolidin-2-one(7)4-methyl-2H-chromen-2-one(26)4-methylisobenzofuran-1(3H)-one(3)4-oxo-4-phenylbutanoic acid(8)4-phenylbutan-2-one(9)4-phenylcyclohexan-1-one(11)4-phenylmorpholin-3-one(6)4-phenylpyrrolidin-2-one(7)5,6-dihydrobenzo[d]thiazol-7(4H)-one(5)5-(aminomethyl)pyrrolidin-2-one(3)5-(hydroxymethyl)pyrrolidin-2-one(7)5-bromoisoindolin-1-one(3)5-bromoisoindoline-1,3-dione(3)5-bromopyrimidin-4(3H)-one(3)5-chloroisoindoline-1,3-dione(3)5-chloropyrimidin-4(3H)-one(4)5-chloropyrimidine-2,4(1H,3H)-dione(3)5-fluoro-3,4-dihydronaphthalen-1(2H)-one(5)5-fluorochroman-4-one(3)5-fluoroindolin-2-one(4)5-fluoroisoindolin-1-one(3)5-fluoropyrimidin-4(3H)-one(3)5-fluoropyrimidine-2,4(1H,3H)-dione(3)5-hydroxy-2-phenyl-4H-chromen-4-one(36)5-hydroxypiperidin-2-one(3)5-methoxy-2-phenyl-4H-chromen-4-one(10)5-methyl-3,4-dihydronaphthalen-1(2H)-one(6)5-methylindolin-2-one(3)5-oxopyrrolidine-2-carboxylic acid(4)6-bromoisoindolin-1-one(4)6-fluorochroman-4-one(5)6-fluoroindoline-2,3-dione(3)6-fluoroisobenzofuran-1(3H)-one(3)6-hydroxy-2,3-dihydro-1H-inden-1-one(3)6-methoxy-2,3-dihydro-1H-inden-1-one(6)6-methoxy-3,4-dihydronaphthalen-1(2H)-one(5)6-methoxyisoindolin-1-one(3)6-methyl-2,3-dihydro-1H-inden-1-one(3)7,8-dihydroquinolin-5(6H)-one(5)7-(diethylamino)-2H-chromen-2-one(8)7-bromo-3,4-dihydronaphthalen-1(2H)-one(8)7-bromochroman-4-one(4)7-bromoindoline-2,3-dione(3)7-bromoquinazolin-4(3H)-one(4)7-chloro-3,4-dihydronaphthalen-1(2H)-one(6)7-fluorochroman-4-one(5)7-fluoroindolin-2-one(3)7-fluoroquinazolin-4(3H)-one(4)7-hydroxy-2H-chromen-2-one(21)7-methoxy-2H-chromen-2-one(11)7-methoxy-3,4-dihydronaphthalen-1(2H)-one(6)7-methoxychroman-4-one(3)7-methylindoline-2,3-dione(7)8-bromochroman-4-one(4)8-fluoro-3,4-dihydronaphthalen-1(2H)-one(5)8-fluoroisoquinolin-1(2H)-one(3)8-methylchroman-4-one(4)acetophenone(508)azepan-2-one(19)benzo[cd]indol-2(1H)-one(5)benzofuran-2(3H)-one(7)chroman-2-one(5)cyclobutanone(24)cyclohex-2-en-1-one(26)cyclohexane-1,3-dione(12)cyclopent-2-en-1-one(21)cyclopentane-1,3-dione(6)dihydro-2H-pyran-2,6(3H)-dione(5)dihydrofuran-2,5-dione(11)dihydrofuran-3(2H)-one(8)dihydrothiophen-3(2H)-one(4)ethene-1,1-diyldibenzene(10)ethyl 2-oxo-2-phenylacetate(10)ethyl 3-oxo-3-phenylpropanoate(24)furan-2(5H)-one(26)isoquinolin-1-ol(8)methyl 3-oxo-3-phenylpropanoate(8)morpholin-3-one(11)phenyl(3-(trifluoromethyl)phenyl)methanone(3)phenyl(m-tolyl)methanone(3)phenyl(p-tolyl)methanone(4)phenyl(pyridin-3-yl)methanone(8)phenyl(thiophen-2-yl)methanone(6)piperidine-2,4-dione(5)propiophenone(38)pyrimidine-4,6(1H,5H)-dione(3)tert-butyl (2-oxopiperidin-3-yl)carbamate(4)tert-butyl (2-oxopyrrolidin-3-yl)carbamate(3)tert-butyl (3-oxocyclohexyl)carbamate(3)tert-butyl (5-oxopyrrolidin-3-yl)carbamate(3)tert-butyl (6-oxopiperidin-3-yl)carbamate(3)tert-butyl 2-oxopiperidine-1-carboxylate(3)tert-butyl 2-oxopyrrolidine-1-carboxylate(15)tert-butyl 4-oxopiperidine-1-carboxylate(16)tetrahydropyrimidin-2(1H)-one(6)2,2,2-trichloro-ethanone(5)2,2-dihydroxy-ethanone(5)2,2-dimethylpropan-1-one(14)2-amino-ethanone(11)2-bromoethanone(106)2-chloroacetyl(39)2-hydroxyethanone(9)2-oxoacetaldehyde(11)2-oxopropanoic acid(3)3-(dimethylamino)prop-2-enoyl(13)3-buten-2-one(10)3-chloro-1-propanone(9)3-oxo-propanenitrile(34)3-oxobutanamide(8)4,4,4-trifluoro-1,3-dione(8)4-chloro-butan-1-one(5)4-oxo-butanoic acid(25)4-oxobut-2-enoic acid(4)5-oxo-pentanoic acid(6)acetylacetonate(26)acetylquinoline(5)benzofuran-one(89)benzoimidazol-one(50)benzophenone(20)benzorophenone(3)benzothiazole-thione(12)benzoxazin-one(50)benzoxazol-one(3)benzoyl(349)bromindan-one(5)bromoethanone(4)butan-1-one(14)butan-2-one(15)butane-1,3-dione(9)butanedione(6)butanedione(6)butanone(10)butanone(10)carbazol-one(14)carbonyl cyanide(4)carbonyl-phenyl-boronic acid(158)carbonyl-phenyl-boronic acid/ester(48)chloro-one(551)chloroethanone(2)chloropropiophenone(1)chroman-one(115)chromanone(4)chromanone(4)chromen-one(38)dicarbonyl(2)dicarbonyl(2)dichloroacetophenone(5)dienone(2)diethanone(5)diethanone(5)difluoroacetophenone(2)dimethyl-indanone(5)dimethylamino-propan-1-one(4)dione(1793)dioxobutanoate(2)dioxohexanoate(10)dioxopentanoate(13)dithione(4)dithione(4)enone(21)ethanone(1026)ethyl 2,4-dioxobutanoate(6)ethyl 2-oxoacetate(21)ethyl 3-oxopropanoate(45)fluoren-one(31)fluorobenzoyl(38)furan-one(251)heptanedionato(7)heptanedionato(7)hexafluoroacetylacetonato(5)hexan-1-one(4)hydroxyethanone(3)imidazolidin-one(34)imidazopyridin-one(25)indanone(26)indazol-one(8)inden-one(164)indol-one(249)indolin-one(189)isonicotinoyl(1)isoquinolin-one(105)methoxy-indanone(37)methyl 2,4-dioxo-butanoate(7)methyl 2-oxoacetate(8)methyl 3-oxopropanoate(16)methyl-pyrazolone(54)morpholinone(4)naphthyridin-one(56)nicotinoyl-chloride(7)oxoacetaldehyde(4)oxoazepane(3)oxoazepane(3)oxochroman(5)oxopropionate(3)oxotetrahydro(11)pentan-1-one(13)phenyl-chromen-one(154)phenyl-one(6)phthalazin-one(17)piperidinone(14)prop-2-en-1-one(13)propan-1-one(78)propan-2-one(32)purin-one(52)pyran-one(165)pyrazin-one(60)pyrazol-one(39)pyrazolopyridin-one(24)pyridazin-one(78)pyridinone(6)pyrimidinone(9)pyrrolidinone(59)pyrrolopyridin-one(63)quinazolin-one(6)quinolinone(3)tetrahydropyran-one(109)tetraone(48)tetraone(48)thiazolidine-one(24)triazine-one(27)triazol-one(48)trifluoro-ethanone(60)trifluoroacetophenone(9)trifluoroethanone(15)trifluoromethyl-acetophenone(50)trifluoromethyl-phenyl-one(23)trione(39)
Nitriles(28341)
1-phenyl-1H-pyrazole-4-carbonitrile(13)1-phenylcyclobutane-1-carbonitrile(6)1-phenylcyclopropane-1-carbonitrile(19)1H-imidazole-4-carbonitrile(2)1H-indazole-3-carbonitrile(9)1H-indole-2-carbonitrile(9)1H-indole-3-carbonitrile(17)1H-indole-5-carbonitrile(5)1H-indole-6-carbonitrile(4)1H-indole-7-carbonitrile(6)1H-pyrazole-4-carbonitrile(5)1H-pyrrole-2-carbonitrile(6)1H-pyrrole-3-carbonitrile(8)2,3-difluorobenzonitrile(17)2,4-dichlorobenzonitrile(4)2,4-difluorobenzonitrile(10)2,6-dibromobenzonitrile(3)2,6-dichlorobenzonitrile(8)2,6-difluorobenzonitrile(21)2-(1,3,2-dioxaborinan-2-yl)benzonitrile(4)2-(2-bromophenyl)acetonitrile(10)2-(2-chlorophenyl)acetonitrile(15)2-(2-fluorophenyl)acetonitrile(15)2-(3-chlorophenyl)acetonitrile(7)2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile(12)2-(4-(benzyloxy)phenyl)acetonitrile(3)2-(bromomethyl)benzonitrile(10)2-(piperazin-1-yl)benzonitrile(6)2-(pyridin-2-yl)acetonitrile(8)2-(pyridin-3-yl)acetonitrile(6)2-(trifluoromethoxy)benzonitrile(5)2-(trifluoromethyl)benzonitrile(26)2-amino-3-bromobenzonitrile(6)2-amino-3-chlorobenzonitrile(3)2-amino-6-chlorobenzonitrile(2)2-aminonicotinonitrile(6)2-bromo-6-fluorobenzonitrile(7)2-chloro-3-methylbenzonitrile(4)2-chloro-4-fluorobenzonitrile(6)2-chloro-6-fluorobenzonitrile(4)2-chloro-N-methylaniline(3)2-chlorobenzonitrile(35)2-hydroxybenzonitrile(21)2-iodobenzonitrile(22)2-naphthonitrile(13)2-nitrobenzonitrile(25)2-oxo-2H-chromene-3-carbonitrile(5)2-phenoxyacetonitrile(8)2-phenoxybenzonitrile(5)2-phenylacetonitrile(73)3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile(16)3-(trifluoromethoxy)benzonitrile(7)3-(trifluoromethyl)benzonitrile(23)3-bromo-2-fluorobenzonitrile(9)3-bromo-2-hydroxybenzonitrile(3)3-bromobenzonitrile(36)3-chloro-2-fluorobenzonitrile(4)3-chlorobenzonitrile(63)3-chloropyrazine-2-carbonitrile(6)3-fluoropicolinonitrile(5)3-hydroxybenzonitrile(20)3-iodobenzonitrile(15)3-nitrobenzonitrile(31)3-oxo-3-phenylpropanenitrile(11)3-phenoxybenzonitrile(4)4-amino-2-chlorobenzonitrile(8)4-chloropyrimidine-5-carbonitrile(6)4-cyanophenyl benzoate(11)4-cyclohexylbenzonitrile(10)4-fluorobenzonitrile(43)4-nitrobenzonitrile(10)5-fluoropicolinonitrile(5)6-methylnicotinonitrile(7)6-methylpicolinonitrile(10)[1,1'-biphenyl]-2-carbonitrile(11)[1,1'-biphenyl]-4-carbonitrile(22)benzo[b]thiophene-2-carbonitrile(3)benzo[d]thiazole-2-carbonitrile(11)benzonitrile(479)furan-2-carbonitrile(4)imidazo[1,2-a]pyridine-6-carbonitrile(3)isonicotinonitrile(16)isoquinoline-1-carbonitrile(7)methyl 2-cyanobenzoate(10)methyl 3-cyanobenzoate(14)morpholine-2-carbonitrile(4)nicotinonitrile(82)pyrazine-2-carbonitrile(15)pyrimidine-2-carbonitrile(14)pyrimidine-4-carbonitrile(9)pyrimidine-5-carbonitrile(6)pyrrolidine-2-carbonitrile(9)pyrrolidine-3-carbonitrile(6)thiazole-5-carbonitrile(7)thiophene-2-carbonitrile(23)thiophene-3-carbonitrile(13)2-amino-acetonitrile(5)2-cyano-acrylic acid(5)2-hydroxyacetonitrile(5)2-methylpropanenitrile(22)3-oxo-propanenitrile(34)acetonitrile(255)acrylonitrile(8)amino-cyano(5)aminoacetonitrile(2)aminobutanenitrile(5)aminocyclohexanecarbonitrile(6)benzonitrile(360)benzopyran-nitrile(1)benzothiazole-nitrile(24)benzothiophene-nitrile(4)bromobenzonitrile(15)bromonicotinonitrile(9)bromophenylacetonitrile(2)butanenitrile(2)butanenitrile(2)carbonyl cyanide(4)chlorobenzonitrile(27)chloroisonicotinonitrile(4)chloronicotinonitrile(40)chromene-carbonitrile(4)cyanide(434)cyanide(434)cyano-methyl(4)cyanoacetamide(4)cyanoacetate(14)cyanobenzaldehyde(4)cyanobenzoate(27)cyanobenzoic(20)cyanocyclopropanecarboxylate(4)cyanoethyl(15)cyanoethyl(15)cyanoisonicotinate(4)cyanomethoxy(10)cyanomethyl(42)cyanomethyl(42)cyanonicotinate(8)cyanophenoxy(8)cyanophenyl(64)cyanophenylboronic(5)cyanopiperazine(3)cyanopiperidine(14)cyanopropan(4)cyanopropan(4)cyanopropanoate(9)cyanopyrrolidine(8)cyanotetrahydro(50)diacetonitrile(1)diaminobenzonitrile(8)dibromoisonicotinonitrile(3)dibromonicotinonitrile(2)dicarbonitrile(45)dicarbonitrile(45)dichlorobenzonitrile(12)dichloroisonicotinonitrile(4)difluorobenzonitrile(41)enecarbonitrile(8)ethyl cyano-carboxylate(8)fluorobenzonitrile(85)fluoroisonicotinonitrile(5)fluoroisophthalonitrile(3)fluoronicotinonitrile(14)fluorophenylacetonitrile(8)fluoropicolinonitrile(13)formylnicotinonitrile(5)formylnicotinonitrile(5)hydrazinylbenzonitrile(6)imidazolyl-nitrile(37)imidazopyridine-carbonitrile(7)indazole-carbonitrile(25)indene-carbonitrile(14)indole-carbonitrile(33)indoline-nitrile(4)iodobenzonitrile(44)iodonicotinonitrile(8)malononitrile(43)malononitrile(43)methoxyisonicotinonitrile(7)methoxyisonicotinonitrile(7)methoxynicotinonitrile(18)methoxynicotinonitrile(18)methylbutanenitrile(7)methylbutanenitrile(7)methylnicotinonitrile(57)methylnicotinonitrile(57)methylpropanenitrile(9)methylpropanenitrile(9)morpholine-nitrile(26)nitropicolinonitrile(10)oxopentanenitrile(7)oxopentanenitrile(7)oxopropanenitrile(8)phenyl-nitrile(119)phenylacrylonitrile(21)propanenitrile(41)propanenitrile(41)pyrazino-nitrile(23)pyrazole-carbonitrile(37)pyrazolopyridine-carbonitrile(16)pyrimidinecarbonitrile(11)pyrrole-carbonitrile(67)pyrrolopyridine-carbonitrile(24)quinoline-carbonitrile(94)tetracarbonitrile(4)tetracarbonitrile(4)tetrafluorobenzonitrile(5)tetrahydroquinoline-carbonitrile(3)thiazole-nitrile(52)thiocyanato(4)thiocyanatoaniline(3)thiophenecarbonitrile(3)thiopheno-nitrile(38)triazole-carbonitrile(5)tricarbonitrile(5)tricarbonitrile(5)trifluorobenzonitrile(7)trifluoromethylbenzonitrile(5)
Nitroes(23612)
(2-nitrophenyl)boronic acid(9)(2-nitrophenyl)methanol(17)(2-nitrovinyl)benzene(16)(3-nitrophenyl)(phenyl)methanone(5)(3-nitrophenyl)boronic acid(17)(3-nitrophenyl)methanol(16)(4-nitrophenyl)(phenyl)methanone(3)(4-nitrophenyl)methanol(12)1,2-dichloro-4-nitrobenzene(12)1,2-difluoro-3-nitrobenzene(15)1,2-difluoro-4-nitrobenzene(20)1,2-dimethyl-3-nitrobenzene(4)1,2-dinitrobenzene(4)1,3-dibromo-2-nitrobenzene(3)1,3-dichloro-2-nitrobenzene(5)1,3-difluoro-2-nitrobenzene(10)1,3-dimethyl-2-nitrobenzene(8)1,3-dinitrobenzene(24)1,4-difluoro-2-nitrobenzene(17)1,4-dimethyl-2-nitrobenzene(7)1-(2-nitrophenyl)ethan-1-one(13)1-(3-methoxy-2-nitrophenyl)ethan-1-one(4)1-(3-nitrophenyl)ethan-1-one(13)1-(3-nitrophenyl)propan-1-one(4)1-(benzyloxy)-2-nitrobenzene(3)1-(benzyloxy)-3-nitrobenzene(10)1-(bromomethyl)-2-nitrobenzene(21)1-(methylsulfonyl)-3-nitrobenzene(6)1-(tert-butyl)-2-nitrobenzene(4)1-(tert-butyl)-3-nitrobenzene(5)1-bromo-2-chloro-4-nitrobenzene(4)1-bromo-2-fluoro-3-nitrobenzene(10)1-bromo-2-fluoro-4-nitrobenzene(6)1-bromo-3-methyl-2-nitrobenzene(3)1-bromo-3-nitrobenzene(98)1-chloro-2-fluoro-4-nitrobenzene(6)1-chloro-2-methyl-3-nitrobenzene(5)1-chloro-2-nitrobenzene(60)1-chloro-3-fluoro-2-nitrobenzene(5)1-chloro-3-nitrobenzene(66)1-ethoxy-3-nitrobenzene(5)1-fluoro-3-nitrobenzene(88)1-iodo-2-nitrobenzene(32)1-iodo-3-nitrobenzene(24)1-methoxy-3-nitrobenzene(59)1-methoxy-4-nitrobenzene(38)1-methyl-2-nitrobenzene(76)1-methyl-3-nitrobenzene(65)1-methyl-4-nitrobenzene(35)1-nitro-2-(trifluoromethoxy)benzene(4)1-nitro-2-(trifluoromethyl)benzene(28)1-nitro-2-phenoxybenzene(4)1-nitro-3-(trifluoromethoxy)benzene(5)1-nitro-3-(trifluoromethyl)benzene(50)1-nitro-4-phenoxybenzene(5)1-nitronaphthalene(3)2,4-dichloro-1-nitrobenzene(15)2,4-difluoro-1-nitrobenzene(29)2,4-dimethoxy-1-nitrobenzene(4)2,4-dimethyl-1-nitrobenzene(10)2-((2-nitrophenyl)amino)ethan-1-ol(4)2-(2-nitrophenyl)acetic acid(15)2-(4-nitrophenyl)acetic acid(7)2-bromo-1-chloro-4-nitrobenzene(7)2-bromo-1-fluoro-3-nitrobenzene(6)2-bromo-1-fluoro-4-nitrobenzene(17)2-bromo-1-methyl-4-nitrobenzene(7)2-bromo-3-nitrobenzoic acid(3)2-bromo-4-chloro-1-nitrobenzene(4)2-bromo-4-fluoro-1-nitrobenzene(14)2-bromo-4-nitroaniline(8)2-bromo-6-nitroaniline(14)2-bromo-6-nitrophenol(9)2-chloro-1,3-dinitrobenzene(2)2-chloro-1-fluoro-4-nitrobenzene(11)2-chloro-1-methyl-3-nitrobenzene(3)2-chloro-3-nitrobenzoic acid(4)2-chloro-4-fluoro-1-nitrobenzene(9)2-chloro-4-methyl-1-nitrobenzene(6)2-chloro-4-nitrophenol(4)2-chloro-5-nitropyrimidine(5)2-chloro-6-nitroaniline(10)2-chloro-6-nitrophenol(6)2-fluoro-1-methoxy-4-nitrobenzene(6)2-fluoro-4-methyl-1-nitrobenzene(4)2-iodo-1-methyl-4-nitrobenzene(3)2-iodo-6-nitroaniline(3)2-methoxy-5-nitropyridine(8)2-methoxy-6-nitrophenol(5)2-methyl-3-nitrobenzoic acid(5)2-methyl-3-nitropyridine(9)2-methyl-6-nitroaniline(6)2-nitro-1H-pyrrole(3)2-nitro-N-phenylaniline(3)2-nitrobenzaldehyde(33)2-nitrobenzoic acid(47)2-nitrobenzonitrile(25)2-nitrofuran(8)2-nitronaphthalene(4)2-nitropyridine(40)2-nitrothiophene(20)3-chloro-2-nitroaniline(4)3-chloro-4-nitroaniline(4)3-fluoro-4-nitroaniline(6)3-methoxy-2-nitrobenzaldehyde(3)3-methoxy-4-nitrobenzoic acid(3)3-methyl-5-nitropyridine(4)3-nitroaniline(55)3-nitrobenzaldehyde(28)3-nitrobenzamide(11)3-nitrobenzene-1,2-diol(6)3-nitrobenzenesulfonamide(8)3-nitrobenzoic acid(61)3-nitrobenzonitrile(31)3-nitrophenol(42)3-nitropyridin-2-ol(7)3-nitropyridin-4-amine(5)3-nitropyridine(54)3-nitrothiophene(6)4,4,5,5-tetramethyl-2-(2-nitrophenyl)-1,3,2-dioxaborolane(5)4-fluoro-1-methyl-2-nitrobenzene(7)4-fluoro-2-nitroaniline(9)4-hydroxy-3-nitrobenzaldehyde(5)4-methyl-3-nitroaniline(4)4-methyl-3-nitropyridine(8)4-nitro-1H-indazole(10)4-nitro-1H-indole(6)4-nitro-1H-pyrazole(8)4-nitroaniline(21)4-nitrobenzaldehyde(11)4-nitrobenzene-1,3-diol(4)4-nitrobenzoic acid(17)4-nitrobenzonitrile(10)4-nitrophenol(36)4-nitropyridine(11)5-(4-nitrophenyl)oxazole(3)5-methoxy-2-nitroaniline(5)5-methyl-2-nitroaniline(4)5-nitro-1H-indazole(5)5-nitro-1H-indole(6)5-nitro-1H-pyrazole(9)5-nitro-1H-pyrazolo[3,4-b]pyridine(3)5-nitro-2,3-dihydrobenzofuran(3)5-nitrobenzo[d][1,3]dioxole(4)5-nitrobenzo[d]thiazole(3)5-nitroisoquinoline(7)5-nitronicotinic acid(5)5-nitropyridin-2-amine(6)6-nitro-1,2,3,4-tetrahydroisoquinoline(3)6-nitro-1H-indazole(9)6-nitro-1H-indole(8)6-nitrobenzo[d]thiazole(3)7-nitro-1,2,3,4-tetrahydroisoquinoline(3)7-nitro-1H-indazole(11)7-nitro-1H-indole(11)ethyl 2-nitrobenzoate(7)ethyl 3-nitrobenzoate(14)ethyl cinnamate(18)methyl 2-nitrobenzoate(36)methyl 3-nitrobenzoate(51)methyl 4-amino-3-nitrobenzoate(3)methyl 4-nitrobenzoate(15)N-(2-nitrophenyl)acetamide(6)N-methyl-2-nitroaniline(13)N-methyl-4-nitroaniline(3)nitrobenzene(987)(E)-(2-nitrovinyl)(23)bromo-fluoro-nitro-benzene(23)bromo-nitro-indazole(12)bromo-nitrobenzene(188)chloro-nitrobenzene(40)difluoronitrobenzene(9)dinitro(7)dinitroaniline(5)dinitrobenzamide(3)ethynyl-nitrobenzene(9)fluoro-iodo-nitro-benzene(13)fluoromethoxy-nitro-benzene(50)fluoronitrobenzene(14)iodonitrobenzene(35)methyl-nitro-imidazole(44)methyl-nitro-indazole(25)methyl-nitro-indole(5)methyl-nitro-pyrazole(49)nitro-benzooxazin(9)nitro-benzoyl-chloride(11)nitro-dihydroxybenzene(4)nitro-imidazole-carboxylate(6)nitro-indazol-amine(3)nitro-indole-carboxylate(18)nitro-pyrazole-carboxylate(6)nitro-pyrrolopyridine(15)nitro-triazole(2)nitro-trifluoromethyl-benzene(31)nitro-trifluoromethyl-pyridin(7)nitroacetophenone(7)nitroaniline(211)nitroanisole(13)nitrobenzaldehyde(100)nitrobenzamide(35)nitrobenzene(295)nitrobenzenesulfonamide(26)nitrobenzenesulfonate(3)nitrobenzoate(153)nitrobenzofuran(8)nitrobenzonitrile(106)nitrobenzotrifluoride(16)nitroimidazole(19)nitroisonicotinate(2)nitroisophthalate(4)nitronicotinate(8)nitroprop(1)nitroprop(1)nitropyrimidine(24)nitropyrrole-carboxylate(5)nitroquinoline(76)nitroquinoxaline(8)nitroso(3)tetrahydroisoquinoline-nitro(6)tetrahydroquinoline-nitro(9)
Phenols(4337)
1,2,3,4-tetrahydroisoquinolin-5-ol(3)1,2,3,4-tetrahydroisoquinolin-6-ol(6)1-(2-hydroxyphenyl)ethan-1-one(53)1-(3-hydroxyphenyl)ethan-1-one(14)1-hydroxyanthracene-9,10-dione(12)2,3-dichlorophenol(6)2,3-difluorophenol(33)2,4-dibromophenol(9)2,4-difluorophenol(18)2,5-dichlorophenol(5)2,6-dibromophenol(17)2,6-dichlorophenol(12)2,6-difluorophenol(18)2,6-diiodophenol(16)2,6-dimethylphenol(13)2-(2-hydroxyphenyl)acetic acid(5)2-(3-hydroxyphenyl)acetic acid(4)2-(benzyloxy)phenol(5)2-(difluoromethoxy)phenol(5)2-(tert-butyl)phenol(38)2-(trifluoromethoxy)phenol(6)2-(trifluoromethyl)phenol(18)2-amino-3-bromophenol(3)2-amino-3-chlorophenol(4)2-aminophenol(57)2-benzylphenol(8)2-bromo-3-methylphenol(4)2-bromo-6-methylphenol(7)2-bromobenzene-1,3-diol(7)2-bromobenzene-1,4-diol(3)2-chloro-4-fluorophenol(9)2-chloro-4-nitrophenol(4)2-chloro-6-methylphenol(4)2-chloro-6-nitrophenol(6)2-chlorobenzene-1,3-diol(4)2-chlorophenol(68)2-ethylphenol(6)2-hydroxy-N-phenylbenzamide(3)2-hydroxybenzamide(11)2-hydroxybenzonitrile(21)2-iodobenzene-1,3-diol(3)2-iodophenol(52)2-isopropylphenol(17)2-methoxyphenol(134)3,4-dibromophenol(3)3,4-difluorophenol(13)3-(benzyloxy)phenol(5)3-(trifluoromethoxy)phenol(8)3-(trifluoromethyl)phenol(24)3-bromo-2-fluorophenol(5)3-bromo-4-chlorophenol(4)3-bromo-4-fluorophenol(10)3-bromobenzene-1,2-diol(8)3-chloro-4-hydroxybenzaldehyde(4)3-chlorobenzene-1,2-diol(6)3-chlorophenol(58)3-fluorophenol(92)3-hydroxybenzonitrile(20)3-iodophenol(13)3-nitrobenzene-1,2-diol(6)3-nitrophenol(42)4-(benzyloxy)phenol(3)4-(trifluoromethyl)phenol(5)4-amino-2-fluorophenol(7)4-amino-3-chlorophenol(4)4-benzylphenol(14)4-chlorobenzene-1,3-diol(3)4-fluorophenol(63)4-hydroxybenzenesulfonic acid(3)4-nitrobenzene-1,3-diol(4)4-nitrophenol(36)6-hydroxy-2,3-dihydro-1H-inden-1-one(3)ethyl 2-hydroxybenzoate(11)ethyl 3-hydroxybenzoate(7)m-cresol(76)methyl 2-(4-hydroxyphenyl)acetate(6)methyl 2-hydroxybenzoate(39)methyl 3-hydroxybenzoate(31)o-cresol(95)p-cresol(39)pyrocatechol(96)resorcinol(94)aminophenol(9)bromophenol(13)chlorophenol(28)dibromonaphtho(5)dibromophenol(9)dichlorophenol(17)difluorophenol(36)dihydroxybenzaldehyde(16)dihydroxybenzamide(6)dihydroxybenzoate(15)dihydroxybenzoic(17)dihydroxybenzonitrile(6)dihydroxyphenethyl(2)dihydroxyphenyl(98)dihydroxystyryl(4)diiodophenol(7)fluorophenol(73)hydroxyacetophenone(16)hydroxybenzaldehyde(88)hydroxybenzamide(24)hydroxybenzenesulfonic(4)hydroxybenzenesulfonic acid(1)hydroxybenzoate(112)hydroxybenzohydrazide(4)hydroxybenzoic(89)hydroxybenzonitrile(60)hydroxybenzoyl(1)hydroxyisophthalate(2)hydroxyphenoxy(7)hydroxyphenylacetic(7)hydroxyphenylacetic acid(5)hydroxyphenylboronic(8)hydroxypropiophenone(3)iodophenol(41)nitrophenol(134)tetrafluorophenol(9)trichlorophenol(4)trifluorophenol(5)trihydroxybenzaldehyde(5)trihydroxybenzoate(12)trihydroxybenzoic(5)trihydroxybenzoic acid(4)trihydroxyphenyl(10)
Phosphoruses(801)
1,1,1-trifluoro-N-(4-oxido-2,6-diphenyldinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin-4-yl)methanesulfonamide(16)diethyl benzylphosphonate(13)dimethyl(phenyl)phosphine oxide(19)ethyltriphenylphosphonium(2)methyltriphenylphosphonium(3)phenyl phosphate(5)phenylphosphonic acid(6)di-tert-butylphosphine(24)diethoxyphosphoryl(5)diethyl methy phosphonate(19)dihydrogen phosphate(22)dihydrogentriphosphate(11)diisopropylphosphoramidite(4)dimethoxyphosphoryl(4)dimethoxyphosphoryl(4)dimethylphosphine(28)dimethylphosphoryl(55)dioxaphosphepine(146)diphosphate(12)diphosphonic(9)diyldiphosphonic(1)diyldiphosphonic(1)phosphonic acid(10)phosphoramidite(18)phosphorothioate(1)trifluoromethyl-phenyl-phosphino(4)triphenylphosphine(95)
Silicon compounds(363)
1-(triisopropylsilyl)-1H-pyrrolo[2,3-b]pyridine(8)tert-butyldimethyl(phenoxy)silane(13)tetraphenylsilane(7)trimethoxy(phenyl)silane(5)trimethyl(phenyl)silane(18)trimethyl(phenylethynyl)silane(21)butyldimethylsilyl(94)butyldimethylsilyl(94)butyldimethylsilyloxy(8)dimethylsilane(16)dimethylsilane(16)dimethylsilyl(4)divinyldisiloxane(3)tert-butyldimethylsilyloxy(19)tetramethyldisiloxane(3)tetramethyldisiloxane(3)triethoxysilyl(13)triethoxysilyl(13)triethylsilyl(2)triethylsilyl(2)triisopropylsilyl(22)triisopropylsilyl(22)trimethoxysilane(19)trimethoxysilane(19)trimethylsilane(155)
Sulfamides(10173)
2-fluoro-N-phenylbenzenesulfonamide(5)2-fluorobenzenesulfonamide(8)2-methylbenzenesulfonamide(5)3-nitrobenzenesulfonamide(8)4-amino-N-phenylbenzenesulfonamide(2)4-bromo-N-phenylbenzenesulfonamide(19)4-chloro-N-phenylbenzenesulfonamide(1)4-methoxy-N-phenylbenzenesulfonamide(3)4-methoxybenzenesulfonamide(5)4-methyl-N-phenylbenzenesulfonamide(13)N,N-diethylbenzenesulfonamide(7)N,N-dimethylbenzenesulfonamide(19)N-(2-phenoxyphenyl)methanesulfonamide(4)N-(3-chlorophenyl)benzenesulfonamide(5)N-(o-tolyl)benzenesulfonamide(4)N-(tert-butyl)benzenesulfonamide(13)N-ethylbenzenesulfonamide(7)N-isopropylbenzenesulfonamide(7)N-methyl-N-(4-phenylpyrimidin-2-yl)methanesulfonamide(7)N-methylbenzenesulfonamide(13)N-phenylbenzenesulfonamide(41)N-phenylmethanesulfonamide(32)phenylmethanesulfonamide(10)pyridine-2-sulfonamide(6)dimethylsulfamoyl(5)dimethylsulfamoyl(5)ethylsulfonamido(5)methanesulfonamide(54)methanesulfonamide(54)methylmethanesulfonamide(3)methylmethanesulfonamide(3)methylsulfonamido(4)methylsulfonamido(4)N,N-dimethyl sulfonamide(31)N-(tert-butyl)sulfamoyl(13)N-ethyl-sulfonamide(9)N-isopropyl-sulfonamide(9)N-methylmethanesulfonamide(8)N-methylsulfamoyl(20)phenylsulfonamido(127)sulfinamide(55)sulfinamide(55)
Sulfides(10098)
(S)-2-(phenylthio)tetrahydro-2H-pyran(5)1,2-diphenyldisulfane(20)2-((pyridin-2-ylmethyl)thio)-1H-benzo[d]imidazole(7)2-(methylthio)benzo[d]thiazole(6)2-(methylthio)pyridine(5)2-(methylthio)pyrimidin-4-amine(10)2-(methylthio)pyrimidine(19)2-(methylthio)thiazole(5)2-(propylthio)pyrimidine(5)4,6-dichloro-2-(methylthio)pyrimidine(9)4-chloro-2-(methylthio)pyrimidine(29)4-methyl-2-(methylthio)pyrimidine(7)5-bromo-2-(methylthio)pyrimidine(6)benzyl(phenyl)sulfane(5)ethyl(phenyl)sulfane(7)pentafluoro(phenyl)-l6-sulfane(19)phenyl(trifluoromethyl)sulfane(21)(trifluoromethyl)thio(25)2-thioacetic acid(13)benzoimidazole-thione(4)ethyl thioacetate(2)ethylthio(24)isopropylthio(7)methylthio(432)methylthio-phenyl-boronic-acid(2)propylthio(5)tert-butylthio(5)thianthrene(4)thiocyanato(4)thioethanol(5)thiopropanoic acid(4)thioxanthen(23)trifluoromethyl-thio(7)
Sulfones(3961)
(4-(methylsulfonyl)phenyl)boronic acid(6)(ethylsulfonyl)benzene(7)1-(methylsulfonyl)-3-nitrobenzene(6)1-(methylsulfonyl)piperazine(5)1-(methylsulfonyl)piperidine(6)1-(phenylsulfonyl)-1H-1,2,4-triazole(6)1-(phenylsulfonyl)-1H-imidazole(5)1-(phenylsulfonyl)azetidine(7)1-bromo-2-(methylsulfonyl)benzene(4)1-bromo-4-(phenylsulfonyl)benzene(7)1-fluoro-2-(methylsulfonyl)benzene(3)1-fluoro-3-(methylsulfonyl)benzene(9)1-methyl-3-(methylsulfonyl)benzene(4)2-(methylsulfonyl)aniline(3)2-(methylsulfonyl)pyridine(11)2-(methylsulfonyl)pyrimidine(25)3-(methylsulfonyl)aniline(7)3-(methylsulfonyl)benzoic acid(5)3-(methylsulfonyl)pyridine(5)benzenesulfonyl fluoride(51)tetrahydro-2H-thiopyran 1,1-dioxide(6)tetrahydrothiophene 1,1-dioxide(10)methanesulfonylmethyl(4)methylsulfonyl(221)trifluoromethylsulfonyl(5)
Sulfonic Acids(1238)
2-aminobenzenesulfonic acid(8)2-methylbenzenesulfonic acid(8)3-aminobenzenesulfonic acid(4)4-chlorobenzenesulfonic acid(3)4-hydroxybenzenesulfonic acid(3)naphthalene-2-sulfonic acid(4)pyridine-3-sulfonic acid(6)2-hydroxypropanesulfonic acid(3)aminobenzenesulfonic(4)benzenesulfonate(13)benzenesulfonic(6)bromobenzenesulfonic(7)bromobenzenesulfonic acid(3)disulfonic(9)disulfonic(9)ethanesulfonic(3)hydroxypropanesulfonic(3)hydroxypropanesulfonic(3)methanesulfonate(93)methanesulfonic(2)methanesulfonic(2)methylbenzenesulfonate(144)propane-1-sulfonate(3)propanesulfonic(4)propanesulfonic(4)propanesulfonic acid(1)trifluoromethanesulfonic(9)
Sulfonyl Chlorides(4024)
1H-pyrazole-4-sulfonyl chloride(8)2-bromobenzenesulfonyl chloride(8)2-chlorobenzenesulfonyl chloride(24)2-fluorobenzenesulfonyl chloride(31)2-methoxybenzenesulfonyl chloride(15)2-methylbenzenesulfonyl chloride(22)3-(chlorosulfonyl)benzoic acid(12)3-(trifluoromethyl)benzenesulfonyl chloride(12)3-chlorobenzenesulfonyl chloride(21)3-fluorobenzenesulfonyl chloride(22)4-chlorobenzenesulfonyl chloride(12)4-fluorobenzenesulfonyl chloride(25)isoquinoline-5-sulfonyl chloride(3)methyl 2-(chlorosulfonyl)benzoate(7)phenylmethanesulfonyl chloride(20)benzenesulfonyl-chloride(185)fluoro-benzenesulfonyl-chloride(32)fluorobenzenesulfonyl(12)methanesulfonyl chloride(28)trifluoromethyl-benzene-sulfonyl chloride(19)
Trifluoromethyls(28915)
(2,2,2-trifluoroethoxy)benzene(12)(2-(trifluoromethoxy)phenyl)boronic acid(4)(2-(trifluoromethyl)phenyl)boronic acid(14)(2-(trifluoromethyl)phenyl)methanol(6)(2-fluoro-3-(trifluoromethyl)phenyl)boronic acid(6)(3-(trifluoromethoxy)phenyl)boronic acid(3)(3-(trifluoromethoxy)phenyl)methanol(3)(3-(trifluoromethyl)phenyl)boronic acid(15)(3-(trifluoromethyl)phenyl)methanol(6)(4-(trifluoromethyl)phenyl)boronic acid(11)(perfluoropropane-2,2-diyl)dibenzene(7)(trifluoromethyl)benzene(573)1,3-bis(trifluoromethyl)benzene(46)1-(2-(trifluoromethyl)phenyl)ethan-1-one(7)1-(3-(trifluoromethoxy)phenyl)ethan-1-amine(6)1-(3-(trifluoromethyl)phenyl)ethan-1-amine(13)1-(4-(trifluoromethoxy)phenyl)ethan-1-amine(6)1-(bromomethyl)-2-(trifluoromethyl)benzene(10)1-(bromomethyl)-3-(trifluoromethyl)benzene(12)1-bromo-2-(trifluoromethoxy)benzene(19)1-bromo-2-fluoro-3-(trifluoromethyl)benzene(4)1-bromo-3-(trifluoromethyl)benzene(47)1-chloro-2-(trifluoromethoxy)benzene(18)1-chloro-2-(trifluoromethyl)benzene(37)1-chloro-3-(trifluoromethoxy)benzene(10)1-chloro-3-(trifluoromethyl)benzene(60)1-chloro-4-(trifluoromethoxy)benzene(7)1-fluoro-2-(trifluoromethoxy)benzene(18)1-fluoro-2-(trifluoromethyl)benzene(56)1-fluoro-3-(trifluoromethyl)benzene(68)1-fluoro-4-(trifluoromethoxy)benzene(11)1-fluoro-4-(trifluoromethyl)benzene(27)1-iodo-2-(trifluoromethyl)benzene(12)1-iodo-3-(trifluoromethyl)benzene(17)1-methoxy-2-(trifluoromethyl)benzene(14)1-methoxy-3-(trifluoromethoxy)benzene(5)1-methoxy-3-(trifluoromethyl)benzene(23)1-methoxy-4-(trifluoromethyl)benzene(11)1-methyl-2-(trifluoromethoxy)benzene(3)1-methyl-2-(trifluoromethyl)benzene(35)1-methyl-3-(trifluoromethyl)benzene(9)1-nitro-2-(trifluoromethoxy)benzene(4)1-nitro-2-(trifluoromethyl)benzene(28)1-nitro-3-(trifluoromethoxy)benzene(5)1-nitro-3-(trifluoromethyl)benzene(50)1-phenoxy-3-(trifluoromethyl)benzene(2)1-phenoxy-4-(trifluoromethyl)benzene(2)1-phenyl-3-(trifluoromethyl)-1H-pyrazole(5)2,2,2-trifluoro-1-phenylethan-1-amine(38)2,2,2-trifluoro-1-phenylethan-1-ol(16)2,2,2-trifluoro-1-phenylethan-1-one(31)2-(2,2,2-trifluoroethoxy)pyridine(8)2-(trifluoromethoxy)aniline(15)2-(trifluoromethoxy)benzaldehyde(5)2-(trifluoromethoxy)benzoic acid(4)2-(trifluoromethoxy)benzonitrile(5)2-(trifluoromethoxy)phenol(6)2-(trifluoromethoxy)pyridine(6)2-(trifluoromethyl)-1,1'-biphenyl(8)2-(trifluoromethyl)aniline(44)2-(trifluoromethyl)benzaldehyde(16)2-(trifluoromethyl)benzamide(4)2-(trifluoromethyl)benzoic acid(24)2-(trifluoromethyl)benzonitrile(26)2-(trifluoromethyl)phenol(18)2-(trifluoromethyl)pyrazine(6)2-(trifluoromethyl)pyridine(6)2-(trifluoromethyl)pyrimidine(27)2-(trifluoromethyl)quinazoline(4)2-(trifluoromethyl)thiazole(11)2-(trifluoromethyl)thiophene(11)2-bromo-1-fluoro-4-(trifluoromethyl)benzene(4)2-bromo-4-(trifluoromethyl)aniline(4)2-bromo-6-(trifluoromethyl)aniline(5)2-chloro-3-(trifluoromethyl)aniline(3)2-chloro-4-(trifluoromethyl)pyrimidine(6)2-chloro-6-(trifluoromethyl)aniline(4)2-methoxy-3-(trifluoromethyl)pyridine(6)2-methoxy-5-(trifluoromethyl)pyridine(4)2-phenoxy-5-(trifluoromethyl)pyridine(7)3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5-(trifluoromethyl)pyridine(3)3-(trifluoromethoxy)aniline(12)3-(trifluoromethoxy)benzaldehyde(5)3-(trifluoromethoxy)benzoic acid(8)3-(trifluoromethoxy)benzonitrile(7)3-(trifluoromethoxy)phenol(8)3-(trifluoromethyl)-1H-pyrrolo[2,3-b]pyridine(3)3-(trifluoromethyl)aniline(65)3-(trifluoromethyl)benzaldehyde(20)3-(trifluoromethyl)benzenesulfonyl chloride(12)3-(trifluoromethyl)benzoic acid(21)3-(trifluoromethyl)benzonitrile(23)3-(trifluoromethyl)phenol(24)3-(trifluoromethyl)pyridazine(3)3-(trifluoromethyl)pyridin-2-ol(3)3-(trifluoromethyl)pyridine(52)3-chloro-4-(trifluoromethyl)aniline(5)4,4,4-trifluoro-1-phenylbutane-1,3-dione(10)4,4,5,5-tetramethyl-2-(2-(trifluoromethyl)phenyl)-1,3,2-dioxaborolane(5)4,4,5,5-tetramethyl-2-(3-(trifluoromethoxy)phenyl)-1,3,2-dioxaborolane(5)4-(trifluoromethoxy)aniline(12)4-(trifluoromethyl)-2H-chromen-2-one(6)4-(trifluoromethyl)phenol(5)4-(trifluoromethyl)pyridine(14)4-(trifluoromethyl)pyrimidin-2-amine(6)4-(trifluoromethyl)pyrimidine(12)4-(trifluoromethyl)thiazole(7)4-bromo-3-(trifluoromethyl)aniline(5)5-(trifluoromethyl)pyridin-2-amine(5)5-(trifluoromethyl)pyridin-2-ol(6)6-(trifluoromethyl)-1H-indazole(6)6-(trifluoromethyl)-1H-indole(5)6-(trifluoromethyl)pyridin-2-amine(11)6-(trifluoromethyl)pyridin-3-amine(5)methyl 2-(trifluoromethyl)benzoate(12)methyl 3-(trifluoromethyl)benzoate(10)N-phenyl-4-(trifluoromethyl)aniline(3)phenyl(3-(trifluoromethyl)phenyl)methanone(3)phenyl(trifluoromethyl)sulfane(21)(R)-2,2,2-trifluoro-1-ethanamine(12)(S)-2,2,2-trifluoro-1-ethanamine(13)(trifluoromethyl)thio(25)1,1,1,3,3,3-hexafluoropropan-2-ol(7)2,2,2-trifluoro ethanamine(25)2,2,2-trifluoro ethanol(24)2,2,2-trifluoroacetamido(5)2,2,2-trifluoroethoxy(44)2,2,2-trifluoroethyl(22)4,4,4-trifluoro-1,3-dione(8)bistrifluoromethyl(35)bromo-trifluoromethyl-pyrazole(7)bromo-trifluoromethyl-pyridine(36)bromo-trifluoromethyl-pyrimidine(15)bromo-trifluoromethyl-quinoline(35)bromobenzotrifluoride(2)chloro-iodo-trifluoromethyl-pyridine(7)chloro-trifluoromethyl-pyridin-amine(28)chloro-trifluoromethyl-pyridine(145)chloro-trifluoromethyl-pyrimidine(40)chloro-trifluoromethyl-quinoline(16)chlorobenzotrifluoride(3)fluoro-trifluoromethyl-pyridine(606)fluorobenzotrifluoride(13)hydroxy-trifluoromethyl(8)iodo-trifluoromethyl-pyridine(24)iodobenzotrifluoride(6)methoxy-trifluoromethyl-benzene(22)methoxy-trifluoromethyl-pyridine(37)nitro-trifluoromethyl-benzene(31)nitro-trifluoromethyl-pyridin(7)trifluoro-ethanone(60)trifluoroacetic(2)trifluoromethanamine(1)trifluoromethyl-acetophenone(50)trifluoromethyl-amine(6)trifluoromethyl-benzene-sulfonyl chloride(19)trifluoromethyl-benzimidazole(4)trifluoromethyl-benzothiazole(17)trifluoromethyl-benzothiophene(3)trifluoromethyl-benzoxazole(1)trifluoromethyl-benzyl(83)trifluoromethyl-chroman(3)trifluoromethyl-cinnamic-acid(3)trifluoromethyl-furan(23)trifluoromethyl-hydroxy-quinoline(14)trifluoromethyl-imidazole(35)trifluoromethyl-imidazopyridine(30)trifluoromethyl-indazole(11)trifluoromethyl-indole(47)trifluoromethyl-indoline(18)trifluoromethyl-isoquinoline(7)trifluoromethyl-nicotinate(4)trifluoromethyl-nicotinic acid(21)trifluoromethyl-oxazole(61)trifluoromethyl-phenyl-acetate(15)trifluoromethyl-phenyl-boronic acid(82)trifluoromethyl-phenyl-ethanamine(29)trifluoromethyl-phenyl-ethanol(22)trifluoromethyl-phenyl-ethylamine(1)trifluoromethyl-phenyl-hydrazine(4)trifluoromethyl-phenyl-hydrochloride(71)trifluoromethyl-phenyl-methanamine(16)trifluoromethyl-phenyl-methanol(29)trifluoromethyl-phenyl-morpholine(13)trifluoromethyl-phenyl-one(23)trifluoromethyl-phenyl-phosphino(4)trifluoromethyl-phenyl-piperazin(23)trifluoromethyl-phenyl-piperidine(28)trifluoromethyl-phenyl-pyrazin(1)trifluoromethyl-phenyl-pyrimidin(35)trifluoromethyl-phenyl-pyrrolidine(5)trifluoromethyl-phenyl-thiazole(3)trifluoromethyl-phenyl-urea(2)trifluoromethyl-picolinamide(7)trifluoromethyl-piperazine(46)trifluoromethyl-piperidine(91)trifluoromethyl-pyrazine(39)trifluoromethyl-pyrazol-amide(24)trifluoromethyl-pyrazole(36)trifluoromethyl-pyrazolone(3)trifluoromethyl-pyridazine(23)trifluoromethyl-pyridin-amine(133)trifluoromethyl-pyridin-methanol(12)trifluoromethyl-pyridine-ethylamine(4)trifluoromethyl-pyrimidin-amine(59)trifluoromethyl-pyrimidin-ol(4)trifluoromethyl-pyrrolo(26)trifluoromethyl-quinazoline(17)trifluoromethyl-quinolin-amine(33)trifluoromethyl-quinoxaline(2)trifluoromethyl-sulfonyl(107)trifluoromethyl-tetrahydroisoquinoline(4)trifluoromethyl-thiazol(72)trifluoromethyl-thio(7)trifluoromethyl-thiophene(34)trifluoromethylaniline(3)trifluoromethylphenol(52)trifluoromethylphenyl(16)trifluoromethylpyridine(6)trifluoromethylpyrimidine(2)trifluoromethylquinoline(173)trifluoromethylsulfonyl(5)trifluorotoluene-dioxaborane(5)
Organometallic Reagents
Organoboron(34718)
(1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid(17)(1-phenyl-1H-pyrazol-4-yl)boronic acid(4)(1H-indazol-4-yl)boronic acid(3)(1H-indazol-6-yl)boronic acid(4)(1H-indol-2-yl)boronic acid(3)(2,3-dichlorophenyl)boronic acid(6)(2,3-difluorophenyl)boronic acid(16)(2,6-dichlorophenyl)boronic acid(5)(2,6-difluorophenyl)boronic acid(13)(2-(phenoxymethyl)phenyl)boronic acid(3)(2-(trifluoromethoxy)phenyl)boronic acid(4)(2-(trifluoromethyl)phenyl)boronic acid(14)(2-bromophenyl)boronic acid(31)(2-chloro-3-methoxyphenyl)boronic acid(4)(2-chloro-6-fluorophenyl)boronic acid(6)(2-fluoro-3-(trifluoromethyl)phenyl)boronic acid(6)(2-fluorophenyl)boronic acid(72)(2-nitrophenyl)boronic acid(9)(3,4-dichlorophenyl)boronic acid(6)(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol(7)(3-(benzyloxy)phenyl)boronic acid(9)(3-(phenoxymethyl)phenyl)boronic acid(3)(3-(phenylcarbamoyl)phenyl)boronic acid(3)(3-(trifluoromethoxy)phenyl)boronic acid(3)(3-(trifluoromethyl)phenyl)boronic acid(15)(3-bromo-2-fluorophenyl)boronic acid(7)(3-bromo-2-methoxyphenyl)boronic acid(6)(3-bromophenyl)boronic acid(25)(3-chloro-2-methoxyphenyl)boronic acid(4)(3-chloro-4-fluorophenyl)boronic acid(7)(3-chlorophenyl)boronic acid(56)(3-fluorophenyl)boronic acid(96)(3-iodophenyl)boronic acid(4)(3-nitrophenyl)boronic acid(17)(4-(benzyloxy)phenyl)boronic acid(12)(4-(methylsulfonyl)phenyl)boronic acid(6)(4-(phenoxymethyl)phenyl)boronic acid(6)(4-(phenylcarbamoyl)phenyl)boronic acid(9)(4-(trifluoromethyl)phenyl)boronic acid(11)(4-cyclohexylphenyl)boronic acid(7)(4-fluorophenyl)boronic acid(43)(5-(trifluoromethyl)pyridin-3-yl)boronic acid(4)(6-phenoxypyridin-3-yl)boronic acid(5)(9H-fluoren-2-yl)boronic acid(5)(E)-4,4,5,5-tetramethyl-2-styryl-1,3,2-dioxaborolane(16)1,3,2-dioxaborinane(6)1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl)piperazine(5)1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)piperazine(10)1-(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine(6)1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole(6)2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-fluorene(7)2-(1,3,2-dioxaborinan-2-yl)benzonitrile(4)2-(1,3,2-dioxaborolan-2-yl)-1-methyl-1H-indole(6)2-(2-(bromomethyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(9)2-(2-bromophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(5)2-(3-chlorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(8)2-(3-cyclopropylphenyl)-1,3,2-dioxaborolane(5)2-(3-isopropoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(5)2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile(12)2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol(7)2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(6)2-(4-(benzyloxy)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(10)2-(4-bromophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(8)2-(4-cyclopropylphenyl)-1,3,2-dioxaborolane(8)2-(4-fluorophenyl)-1,3,2-dioxaborinane(5)2-([1,1'-biphenyl]-4-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(10)2-(cyclohex-1-en-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(16)2-(furan-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(6)2-(m-tolyl)-1,3,2-dioxaborinane(4)2-benzyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(12)2-chloro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(4)2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(14)2-chloro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(6)2-fluoro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(7)2-methoxy-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(7)2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(5)2-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(5)2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(3)3-(1,3,2-dioxaborinan-2-yl)aniline(3)3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile(16)3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(74)3-chloro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(5)3-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(7)3-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(6)4,4,5,5-tetramethyl-2-(2-(trifluoromethyl)phenyl)-1,3,2-dioxaborolane(5)4,4,5,5-tetramethyl-2-(2-nitrophenyl)-1,3,2-dioxaborolane(5)4,4,5,5-tetramethyl-2-(3-(trifluoromethoxy)phenyl)-1,3,2-dioxaborolane(5)4,4,5,5-tetramethyl-2-(naphthalen-1-yl)-1,3,2-dioxaborolane(19)4,4,5,5-tetramethyl-2-(naphthalen-2-yl)-1,3,2-dioxaborolane(12)4,4,5,5-tetramethyl-2-(thiophen-2-yl)-1,3,2-dioxaborolane(23)4,4,5,5-tetramethyl-2-(thiophen-3-yl)-1,3,2-dioxaborolane(10)4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2,3,6-tetrahydropyridine(11)4-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine(5)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole(5)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indoline(5)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)nicotinonitrile(4)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-amine(5)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine(27)5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiazole(7)[1,1'-biphenyl]-4-ylboronic acid(14)benzo[b]thiophen-2-ylboronic acid(5)benzofuran-2-ylboronic acid(8)benzyl (4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)carbamate(5)cyclohex-1-en-1-ylboronic acid(4)ethyl 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate(6)isoquinolin-4-ylboronic acid(3)methyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)nicotinate(4)N,N-diphenyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline(6)naphthalen-1-ylboronic acid(4)naphthalen-2-ylboronic acid(8)phenylboronic acid(450)pyridin-2-ylboronic acid(9)pyridin-3-ylboronic acid(49)pyridin-4-ylboronic acid(21)pyrimidin-5-ylboronic acid(9)tert-butyl 2-(1,3,2-dioxaborolan-2-yl)-1H-indole-1-carboxylate(6)tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole-1-carboxylate(3)tetraphenylborate(8)thiophen-2-ylboronic acid(20)alkenyl-phenyl-boronic-acid(2)amino-phenyl-boronic-acid(9)bromophenyl-boronate(2)bromopyridine-boronate(4)butoxy-phenyl-boronic-acid(1)chloro-phenyl-boronic-acid(49)dimethyldioxaborane(20)dioxaborolan-carbazole(21)dioxaborolan-indazole(27)dioxaborolan-indole(90)dioxaborolan-methanol(4)dioxaborolan-pyrazole(161)dioxaborolan-pyrimidine(47)dioxaborolane(47)dioxaborolane-benzene(39)dioxaborolane-benzimidazole(3)dioxaborolane-benzoxazole(3)dioxaborolane-indene(5)dioxaborolane-morpholine(33)dioxaborolane-piperazine(40)dioxaborolane-piperidine(4)dioxaborolane-pyrrolidine(3)dioxaborolane-quinoline(29)dioxaborolane-thiazole(1)fluorene-boronic acid(20)fluorene-dioxaborolane(31)fluorene-phenyl-boronic acid(11)fluorene-phenyl-boronic acid/ester(1)fluoro-phenyl-boronic acid(509)hydroxymethyl-phenyl-boronic-acid(12)indazol-boronic acid(36)isopropoxyphenylboronic(4)isopropyl-phenyl-boronic-acid(1)Isoquinolin-boronic-acid(5)methoxy-pyridine-boronate(38)methoxyphenylboronic acid(34)methoxyphenylboronic-acid(12)methylthio-phenyl-boronic-acid(2)phenyl-boronate(28)phenyl-boronic-acid-carboxylate(4)phenyl-naphthalene-boronic acid(9)phenyl-naphthalene-boronic acid/ester(1)phenyl-naphthalene-boronic-acid(2)phenylboronic(72)picoline-boronate(5)potassium methyltrifluoroborate(21)Potassium trifluoroborate(112)pyrazole-phenyl-boronic-acid(3)pyridine-boronate(187)pyridineboronic acid(4)thienyl-benzene-boronic-acid(4)thiophene-boronic acid(70)trifluoromethyl-phenyl-boronic acid(82)trifluorotoluene-dioxaborane(5)
Inhibitors/Agonists
Anti-infection(633)
3CLpro(18)Antibacterial(304)Antifungal(77)Antiparasitic(89)Antiviral(7)CMV(8)Dengue Virus(4)EBV(1)Enterovirus(1)FCoV(1)Flavivirus(6)HBV(16)HCV(35)HIV(85)HIV Integrase(9)HIV Protease(13)HRV(1)HSV(16)Influenza Virus(35)LpxC(1)M2 proton channel(1)NNRTI(5)NRTI(5)Orthopoxvirus(6)PLP(4)Prion(1)Protein Synthesis(2)Reverse Transcriptase(16)RSV(1)SARS-CoV(22)SQLE(2)Viral MTase(1)Virus Protease(3)β-lactamase(4)
Cell Cycle(537)
Antifolate(4)Aurora Kinase(15)BUB(1)Casein Kinase(17)CDK(60)Chk(8)CRM1(2)DHFR(10)DNA/RNA Synthesis(76)DYRK(7)eIF(3)FOXM1(1)G-quadruplex(1)HDAC(39)HSF(2)HSP(15)IRE1(3)Kinesin(6)LIMK(3)Microtubule/Tubulin(44)Mps1(6)Nucleoside Antimetabolite/Analog(166)PAK(3)PDI(1)PLK(12)PPAR(38)RAD51(5)Rho(5)RNA polymerase 2(1)RNA polymerase?1(1)ROCK(14)Sirtuin(16)SRPK(8)Telomerase(2)TOPK(1)Topoisomerase(50)Wee1(3)YB-1(4)
GPCR/G Protein(562)
5-HT Receptor(96)Adenosine Receptor(34)Adrenergic Receptor(43)Angiotensin Receptor(14)Apelin Receptor(2)Bombesin Receptor(1)Bradykinin Receptor(2)cAMP(3)Cannabinoid Receptor(16)CaSR(5)CCR(14)CGRP Receptor(1)Cholecystokinin Receptor(9)CRF Receptor(3)CXCR(17)Dopamine Receptor(31)Endothelin Receptor(13)Epac(2)GHSR(4)GLP Receptor(2)Glucagon Receptor(6)GNRH Receptor(1)GPCR(55)GPR109A(1)GPR119(2)GPR139(3)GPR24(1)GPR35(1)GPR40(1)GPR52(1)GPR84(1)GPR88(4)Guanylate Cyclase(1)HCAR(1)Histamine Receptor(50)LHCGR(21)LPA Receptor(7)LPL Receptor(27)Melanocortin Receptor(4)mGluR(50)MRGPR(2)Neuropeptide Y receptor(1)Neurotensin Receptor(2)Opioid Receptor(13)OXTR(5)P2Y Receptor(11)PAFR(7)PAR(14)Prostaglandin Receptor(40)Ras(34)RGS(2)Sigma Receptor(6)SOS(1)SRPK(8)SSTR(2)TAAR(2)TSH Receptor(2)Urotensin Receptor(2)Vasopressin Receptor(1)β-arrestin(2)
Immunology/Inflammation(386)
AIM2(5)AP-1(5)Arginase(3)CCR(14)CD19(1)CD20(2)CD22(1)CD28(2)CD3(3)CD38(3)CD73(2)Complement System(6)COX(79)CRTH2(6)CTLA-4(2)CXCR(17)FAP(2)FKBP(14)FLAP(2)FPR(2)Glucocorticoid Receptor(6)GM-CSF(1)Histamine Receptor(50)IFNAR(4)IL(16)IL Receptor(2)Immunology & Inflammation Related(15)IRAK(6)Legumain(1)LTR(11)MALT1(2)MIF(2)MyD88(1)NADPH Oxidase(7)NFAT(2)NLR(3)NO Synthesis(2)NOS(34)Nrf2(13)Others-Others(1)PD-1/PD-L1(4)PG Synthase(5)Pyroptosis(1)ROS(63)Scavenger receptor(1)SIK(1)SOD(4)SPHK(8)STING(9)Thrombopoietin Receptor(2)TLR(18)
Membrane Transporter/Ion Channel(253)
AKK1(1)AQP(2)ATP synthase(4)ATPase(10)BCRP(3)Calcium Channel(49)CFTR(14)Chloride Channel(2)CRAC Channel(3)CRM1(2)EAAT(3)ENT(1)GABA Receptor(27)GLUT(2)Glutamate transporter(4)Glycine transporter(4)HCN Channel(1)IBAT(3)MAT(4)MCT(4)Na+/Ca2+ Exchanger(5)Na+/K+ ATPase(2)Na/HCO3?cotransporter(1)NKCC(1)Oct(1)P-gp(7)P2X Receptor(7)Potassium Channel(45)Proton Pump(10)SGLT(8)SLC(3)Sodium Channel(25)TRP Channel(28)URAT1(2)VDAC(1)
Metabolic Enzyme(481)
3-PGDH(1)6PGD(2)ACAT(2)ACC(4)ACMSD(1)Adenosine Deaminase(2)Adenosylhomocysteinase(6)AhR(8)AKR(1)ALDH(6)Aldose Reductase(9)Aminopeptidase(5)Antifolate(4)AO(1)Arginase(3)ATP Citrate Lyase(2)ATP synthase(4)BDK(1)Carbonic Anhydrase(20)Carboxylesterase(9)Carboxypeptidase(4)Cathepsin(12)Ceramidase(1)CETP(3)CPA(3)CSE(1)Decarboxylase(4)Dehydrogenase(17)DGAT(29)DGK(1)E-NTPDase(1)Elastase(7)Enolase(1)Enteropeptidase(1)FABP(4)Factor Xa(4)Farnesyl Transferase(3)FAS(4)Ferroptosis(8)FOXO(1)FPR(2)FTO(1)Fumarate hydratase(1)FXR(13)G0/G1 switch protein 2(1)Galactosidases(1)GCS(1)Glucokinase(4)Glucosidase(18)GLUT(2)Glyoxalase(2)GOAT(1)GST(5)Hexokinase(1)HMGCR(5)HPSE(1)Hydrolase(2)Hydroxylase(8)IDH(2)IDO(10)LDL(4)LDLR(1)Lipase(30)Lipoxygenase(23)LOX(1)LRH-1(1)LXR(3)MAGL(2)MDH(1)MetAP(2)Mitochondrial Electron Transport Chain(2)Mitochondrial Metabolism(13)NAMPT(6)Neprilysin(2)NNMT(1)NPR(9)NQO(2)P450(38)PAI-1(1)PDE(54)PDHK(1)PEPCK(1)PGAM1(1)PHGDH(1)Phospholipase(19)Phosphorylase(5)PKM2(4)Proprotein convertases(2)Pyruvate Kinase(5)RAR/RXR(20)REV-ERB(4)ROR(2)SCD(5)sEH(4)SGK(1)SHMT(2)Steroid Sulfatase(1)TDO(1)TGH(1)Thioredoxin(2)Thrombin(8)Thymidylate Synthase(6)Transferase(14)TrxR(7)Trypanothione reductase(2)VD/VDR(10)Xanthine Oxidase(3)
Neuronal Signaling(592)
5-HT Receptor(96)AAK1(7)AChE(12)AChR(78)Adenosine Kinase(2)Adrenergic Receptor(43)AMPAR(13)Amyloid-β(17)Calcium Channel(49)CaMK(8)Cannabinoid Receptor(16)CGRP Receptor(1)Cholecystokinin Receptor(9)Cholinesterase(25)COMT(4)DAT(5)Dopamine Receptor(31)Enkephalinase(1)FAAH(6)GABA Receptor(27)GluR(26)GPR119(2)GPR139(3)GPR24(1)Histamine Receptor(50)Huntingtin(3)Imidazoline Receptor(3)MAO(24)Melanocortin Receptor(4)mGluR(50)Neuropeptide Y receptor(1)Neurotensin Receptor(2)NGF(2)NK Receptor(7)NMDAR(14)Notch(2)Nurr1(1)Opioid Receptor(13)OX Receptor(9)P2X Receptor(7)Proprotein convertases(2)RAGE(1)Secretase(23)SERT(6)Sigma Receptor(6)SMS(1)SSRIs(2)Substance P(1)TAAR(2)Trk Receptor(13)TRP Channel(28)TTR(2)α-synuclein(1)
Other Inhibitors/Agonists(458)
20-HETE(1)Amino Acids and Amino Acid Derivatives(31)Aminoacyl-tRNA Synthetase(4)Anti-inflammation(30)Anti-tumor(7)Anti-ulcer(1)Antioxidant(30)Bcat(11)c-RET(9)Cancer Stem Cells(1)CBFβ-SMMHC(1)CD20(2)CD52(1)CD73(2)Chelating Agent(3)CRTH2(6)CSE(1)Decarboxylase(4)Dehydrogenase(17)Drug Metabolite(27)ECE(1)Endogenous Metabolite(139)FAP(2)FATP(1)Fungicide(60)Galectin-3(1)GCase(1)Glucocorticoid Receptor(6)GRK(3)Herbicide(13)HIPK2(1)HNF4α(1)Hydroxylase(8)IgE(2)Insecticide(26)LDL(4)Lipase(30)MLKL(2)Molecular Glues(4)MTTP(3)Neuroprotection/neurogenic Agent(1)Nur77(2)Osteoclasts/Bone/Osteogenesis(6)Others(350)OxPhos(2)PAX2(1)PBR(3)PCSK9(2)PDX1(1)Phosphatase(29)Phosphorylase(5)PIN1(1)PKG(4)Platelet(1)PME-1(2)PREP(4)Protein Synthesis(2)Quorum Sensing(24)RUNX(1)SAP(1)SQS(1)SRPK(8)Thioredoxin(2)TNK(2)UGT1A1(1)Urease(2)vanin(1)Vitamin(15)α-amylase(2)
Protein Tyrosine Kinase/RTK(403)
Ack1(2)ALK(11)Bcr-Abl(19)BMX Kinase(1)BRK(1)BTK(16)c-FMS(10)c-Kit(18)c-Met/HGFR(19)c-RET(9)DDR1/DDR2 Receptor(2)DYRK(7)EGFR(68)Ephrin Receptor(7)ErbB(2)FAK(8)FGFR(22)FLT3(13)GAK(412)HER2(10)IGF-1R(3)Insulin Receptor(7)ITK(2)JAK(27)MRCK(1)PDGFR(23)Pyk2(1)SHP2(32)Src(21)Syk(9)TAM Receptor(5)Tie-2(3)Trk Receptor(13)VEGFR(49)
Cancer
Tumor Epigenetics and Gene Regulation(368)
Aminoacyl-tRNA Synthetase(4)ATM/ATR(11)DNA Methyltransferase(7)DNA/RNA Synthesis(76)Epigenetic Reader Domain(38)HDAC(39)HIF(18)Histone Acetyltransferase(11)Histone Demethylase(33)Histone Methyltransferase(47)METTL3(6)MicroRNA(3)PAD(2)PARP(21)Pim(7)PKC(30)RNA polymerase?1(1)Sirtuin(16)TET(1)Topoisomerase(50)
Catalysts Ligands
Enzymology
Enzyme Inhibitor(57)
Acetaldehyde Dehydrogenase Inhibitor(2)Acyltransferase Inhibitor(4)Alkaline Phosphatase Inhibitor(2)Angiotensin Converting Enzyme Inhibitor(4)ATP Enzyme Inhibitor(7)Calcineurin Inhibitor(1)Carbonic Acid Dehydratase Inhibitor(2)Caspase Inhibitor(2)Cyclooxygenase (COX) Inhibitor(49)DNA Topoisomerase II Inhibitor(11)Hypoxia Inducible Factor (HIF) Inhibitor(3)Inosine Monophosphate (IMP) Dehydrogenase Inhibitor(10)Leukotriene A4 Hydrolase Inhibitor(3)Lipoxygenase Inhibitor(11)Monoamine Oxidase Inhibitor(4)NADH Peroxidase Inhibitor(2)Pancreatic Lipase Inhibitor(2)Phosphodiesterase Inhibitor(20)Protease Inhibitor(20)Reverse Transcriptase Inhibitor(16)Starch Glucosidase Inhibitor(3)Thromboxane Synthase Inhibitor(3)Topoisomerase I Inhibitor(8)Tyrosine / EGF Receptor Protein Kinase Inhibitor(32)
Fluorescent Probe / Staining
Fluorescent Dye(400)
Acridine Dye(2)Apoptosis Dyes(2)BODIPY(3)Cancer Analysis Dyes(2)Coumarine Dye(13)Cyanine Dye(5)Cytotoxicity Analysis Dyes(5)Endocytosis and pinocytosis Dyes(1)Exosome Dyes(1)Lipid droplet(1)Lysosome Dyes(3)Membrane Dyes(1)Mitochondria(1)Neuroscience Analysis Dyes(4)Nucleic Acid(1)Nucleus Dyes(3)Other Fluorescent Dyes(6)Others(350)PH Indicators(5)Protein(3)ROS Dyes(3)
Natural product
Vesicle
Parkinson's disease and Alzheimer's disease(789)
Akt(30)AMPAR(13)Apoptosis(285)Caspase(28)CDK(60)CREB(2)Dopamine Receptor(31)ERK(19)Glucagon Receptor(6)GPCR(55)GSK-3(21)IL(16)JAK(27)JNK(11)LRRK2(5)MyD88(1)NF-κB(58)Notch(2)p53(27)PI3K(51)PKA(16)RAGE(1)Ras(34)Rho(5)ROCK(14)ROS(63)Secretase(23)STAT(30)STING(9)TLR(18)TNF(4)Tyrosinase(17)Tyrosine / EGF Receptor Protein Kinase Inhibitor(32)Wnt(16)α-synuclein(1)β-catenin(4)