| 81% |
|
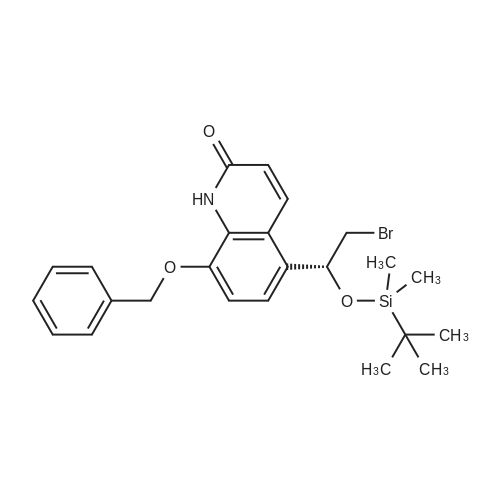
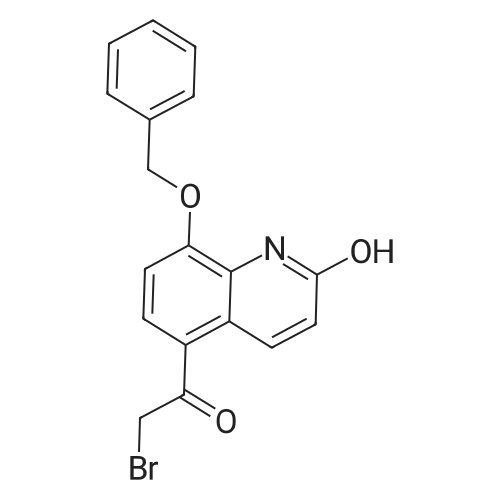
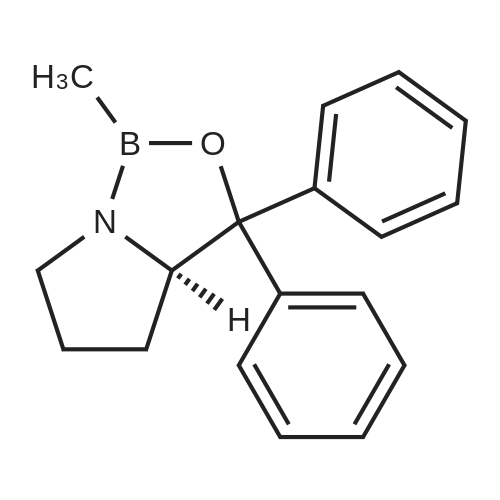
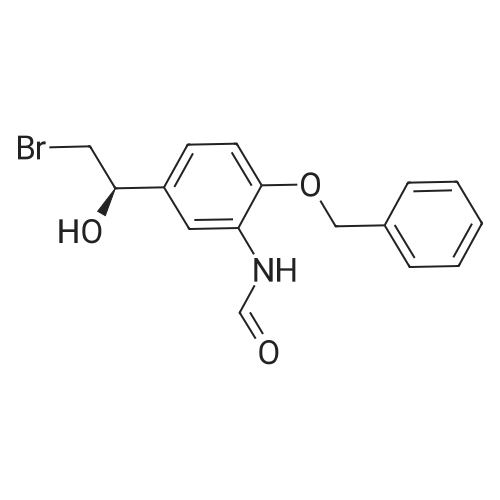
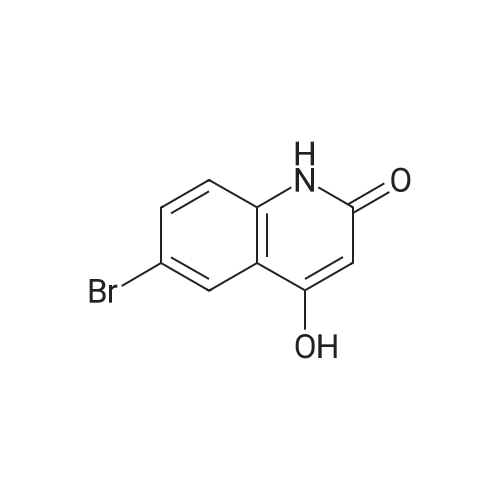
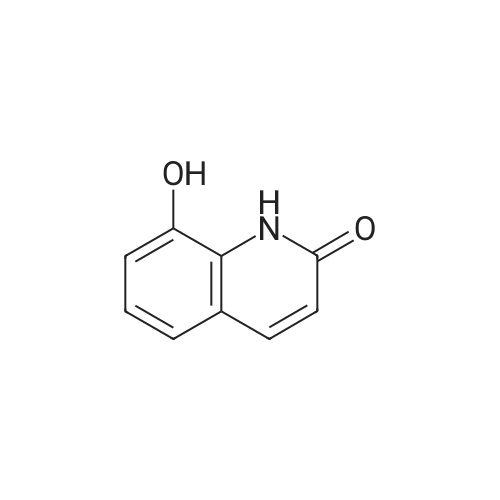
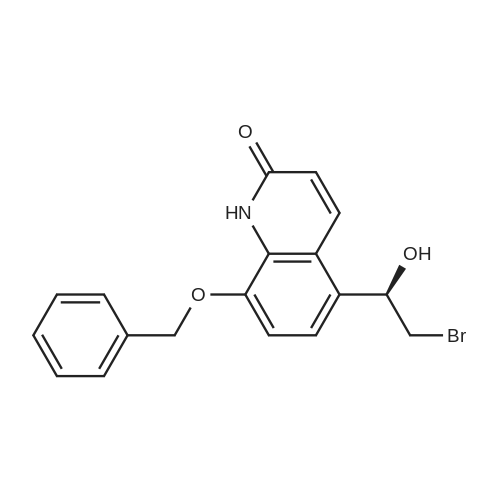
EXAMPLE 2; Synthesis of N-{2-[4-(3-phenyl-4-methoxyphenyl)aminophenyl]ethyl}-(R)-2-hydroxy-2-(8-benzyloxy-2(1H)-quinolinon-5-yl)ethylamine (PP); a. Synthesis of 5-(2-bromo-(R)-1-hydroxy)ethyl-8-benzyloxy-2(1H)-quinolinone (FF); (R)-(+)-alpha,alpha-Diphenylprolinol (30.0 g, 117 mmol) and trimethylboroxine (11.1 ml, 78 mmol) were combined in toluene (300 ml) and stirred at room temperature for 30 minutes. The mixture was placed in a 150 C. oil bath and liquid was distilled off. Toluene was added in 20 ml aliquots, and distillation was continued for 4 hours. A total of 300 ml toluene was added. The mixture was finally cooled to room temperature. A 500 muL aliquot was evaporated to dryness, weighed (246 mg) to determine that the concentration of catalyst was 1.8 M. [0128] 5-(2-Bromo-1-oxy)ethyl-8-benzyloxy-2(1H)-quinolinone (R) (90.0 g, 243 mmol) was placed under nitrogen, tetrahydrofuran (900 mL) was added followed by the catalyst from above (1.8 M in toluene, 15 ml, 27 mmol). The suspension was cooled to -10+5 C. in an ice/isopropanol bath. Borane (1.0 M in THF, 294 ml, 294 mmol) was added over 4 hours. The reaction was stirred an additional 45 minutes at -10 C., then methanol (250 ml) was added slowly. The mixture was concentrated under vacuum. The residue was dissolved in boiling acetonitrile (1.3 l), filtered while hot and cooled to room temperature. The crystals were filtered, washed with acetonitrile and dried under vacuum to give 5-(2-bromo-(R)-1-hydroxy)ethyl-8-benzyloxy-2(1H)-quinolinone (FF) (72.5 g, 196 mmol, 81% yield, 95% ee, 95% pure by HPLC area ratio). |
| 81% |
With borane;(3aR)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2-c][1,3,2]oxazaborole; In tetrahydrofuran; toluene; at -10℃; for 4.75h; |
(R)-(+)-alpha,alpha-Diphenylprolinol (30.0 g, 117 mmol) and trimethylboroxine (11.1 mL, 78 mmol) were combined in toluene (300 mL) and stirred at room temperature for 30 min. The mixture was placed in a 150 C. oil bath and liquid was distilled off. Toluene was added in 20 mL aliquots and distillation was continued for 4 h. A total of 300 mL toluene was added. The mixture was then cooled to room temperature. A 500 muL aliquot was evaporated to dryness and weighed (246 mg) to determine that the concentration of catalyst was 1.8M. [0523] 8-Benzyloxy 5-(2-bromoacetyl)-1H-quinolin-2-one (90.0 g, 243 mmol) was placed under nitrogen and tetrahydrofuran (900 mL) was added followed by the catalyst described above (1.8 M in toluene, 15 mL, 27 mmol). The suspension was cooled to -10+5 C. in an ice/isopropanol bath. Borane (1.0 M in THF, 294 mL, 294 mmol) was added over 4 h. The reaction was then stirred an additional 45 min at -10 C. and then methanol (250 mL) was added slowly. The mixture was concentrated under vacuum and the residue was dissolved in boiling acetonitrile (1.3 L), filtered while hot and then cooled to room temperature. The crystals were filtered, washed with acetonitrile and dried under vacuum to give the title compound (72.5 g, 196 mmol, 81% yield, 95% ee, 95% pure by HPLC). |
| 81% |
|
(b) 8-Benzyloxy-5-((R)-2-bromo-1-hydroxyethyl)-1H-quinolin-2-one (R)-(+)-alpha,alpha-Diphenylprolinol (30.0 g, 117 mmol) and trimethylboroxine (11.1 ML, 78 mmol) were combined in toluene (300 ML) and stirred at room temperature for 30 min.The mixture was placed in a 150 C. oil bath and liquid was distilled off.toluene was added in 20 ML aliquots and distillation was continued for 4 h.A total of 300 ML toluene was added.The mixture was then cooled to room temperature.A 500 muL aliquot was evaporated to dryness and weighed (246 mg) to determine that the concentration of catalyst was 1.8M. 8-Benzyloxy 5-(2-bromoacetyl)-1H-quinolin-2-one (90.0 g, 243 mmol) was placed under nitrogen and tetrahydrofuran (900 ML) was added followed by the catalyst described above (1.8 M in toluene, 15 ML, 27 mmol).The suspension was cooled to -10+5 C. in an ice/isopropanol bath.Borane (1.0 M in THF, 294 ML, 294 mmol) was added over 4 h.The reaction was then stirred an additional 45 min at -10 C. and then methanol (250 ML) was added slowly.The mixture was concentrated under vacuum and the residue was dissolved in boiling acetonitrile (1.3 L), filtered while hot and then cooled to room temperature.The crystals were filtered, washed with acetonitrile and dried under vacuum to give the title compound (72.5 g, 196 mmol, 81% yield, 95% ee, 95% pure by HPLC). |
| 81% |
|
(R)-(+)-alpha,alpha-Diphenylprolinol (30.0 g, 117 mmol) and trimethylboroxine (11.1 mL, 78 mmol) were combined in toluene (300 mL) and stirred at room temperature for 30 min. The mixture was placed in a 150 C. oil bath and liquid was distilled off. Toluene was added in 20 mL aliquots and distillation was continued for 4 h. A total of 300 mL toluene was added. The mixture was then cooled to room temperature. A 500 muL aliquot was evaporated to dryness and weighed (246 mg) to determine that the concentration of catalyst was 1.8 M. 8-Benzyloxy 5-(2-bromoacetyl)-1H-quinolin-2-one (90.0 g, 243 mmol) was placed under nitrogen and tetrahydrofuran (900 mL) was added followed by the catalyst described above (1.8 M in toluene, 15 mL, 27 mmol). The suspension was cooled to -10+-5 C. in an ice/isopropanol bath. Borane (1.0 M in THF, 294 mL, 294 mmol) was added over 4 h. The reaction was then stirred an additional 45 min at -10 C. and then methanol (250 mL) was added slowly. The mixture was concentrated under vacuum and the residue was dissolved in boiling acetonitrile (1.3 L), filtered while hot and then cooled to room temperature. The crystals were filtered, washed with acetonitrile and dried under vacuum to give the title compound (72.5 g, 196 mmol, 81% yield, 95% ee, 95% pure by HPLC). |
| 81% |
|
(7?)-(+)-a,a-Diphenylprolinol (30.0 g, 117 mmol) and trimethylboroxine (11.1 mL, 78 mmol) were combined in toluene (300 mL) and stirred at room temperature for 30 min. The mixture was placed in a 150 C oil bath and liquid was distilled off. Toluene was added in 20 mL aliquots and distillation was continued for 4 h. A total of 300 mL toluene was added. The mixture was then cooled to room temperature. A 500 uL aliquot was evaporated to dryness and weighed (246 mg) to determine that the concentration of catalyst was 1.8 M.8-Benzyloxy 5-(2-bromoacetyl)-l//-quinolin-2-one (90.0 g, 243 mmol) was placed under nitrogen and tetrahydrofuran (900 mL) was added followed by the catalyst described above (1.8 M in toluene, 15 mL, 27 mmol). The suspension was cooled to -10+-5 C in an ice/isopropanol bath. Borane (1.0 M in THF, 294 mL, 294 mmol) was added over 4 h. The reaction was then stirred an additional 45 min at -10 C and then methanol (250 mL) was added slowly. The mixture was concentrated under vacuum and the residue was dissolved in boiling acetonitrile (1.3 L), filtered while hot and then cooled to room temperature. The crystals were filtered, washed with acetonitrile and dried under vacuum to gjve the title compound (72.5 g, 196 mmol, 81% yield, 95% ee, 95% pure by HPLC). |
| 81% |
With borane;(3aR)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2-c][1,3,2]oxazaborole; In tetrahydrofuran; at -10 - 5℃; for 4.75h; |
(R)-(+)-alpha,alpha-Diphenylprolinol (30.0 g, 117 mmol) and trimethylboroxine (11.1 mL, 78 mmol) were combined in toluene (300 mL) and stirred at room temperature for 30 minutes. The mixture was placed in a 150 C. oil bath and liquid was distilled off. Toluene was added in 20 mL aliquots, and distillation was continued for 4 hours. A total of 300 mL toluene was added. The mixture was finally cooled to room temperature. A 500 muL aliquot was evaporated to dryness, weighed (246 mg) to determine that the concentration of catalyst was 1.8 M. 5-(2-Bromo-1-oxy)ethyl-8-benzyloxy-2(1H)-quinolinone (R) (90.0 g, 243 mmol) was placed under nitrogen, tetrahydrofuran (900 mL) was added followed by the catalyst from above (1.8 M in toluene, 15 mL, 27 mmol). The suspension was cooled to -10+/-5 C. in an ice/isopropanol bath. Borane (1.0 M in THF, 294 mL, 294 mmol) was added over 4 hours. The reaction was stirred an additional 45 minutes at -10 C., then methanol (250 mL) was added slowly. The mixture was concentrated under vacuum. The residue was dissolved in boiling acetonitrile (1.3 L), filtered while hot and cooled to room temperature. The crystals were filtered, washed with acetonitrile and dried under reduced pressure to give 5-(2-bromo-(R)-1-hydroxy)ethyl-8-benzyloxy-2(1H)-quinolinone (FF) (72.5 g, 196 mmol, 81% yield, 95% ee, 95% pure by HPLC area ratio). |
| 81% |
|
(R)-(+)-alpha,alpha-DIPHENYLPROLINOL (30.0 g, 117 mmol) and trimethylboroxine (11.1 mL, 78 mmol) were combined in toluene (300 mL) and stirred at room temperature for 30 min. The mixture was placed in a 150 C oil bath and liquid was distilled off. Toluene was added in 20 mL aliquots and distillation was continued for 4 h. A total of 300 mL toluene was added. The mixture was then cooled to room temperature. A 500 pL aliquot was evaporated to dryness and weighed (246 mg) to determine that the concentration of catalyst was 1. 8 M. 8-BENZYLOXY 5- (2-bromoacetyl)-lH-quinolin-2-one (90.0 g, 243 mmol) was placed under nitrogen and tetrahydrofuran (900 mL) was added followed by the catalyst described above (1. 8 M in toluene, 15 mL, 27 mmol). The suspension was cooled TO-1 C in an ice/isopropanol bath. Borane (1.0 M in THF, 294 mL, 294 mmol) was added over 4 h. The reaction was then stirred an additional 45 min at-10 C and then methanol (250 mL) was added slowly. The mixture was concentrated under vacuum and the residue was dissolved in boiling acetonitrile (1.3 L), filtered while hot and then cooled to room temperature. The crystals were filtered, washed with acetonitrile and dried under vacuum to give the title compound (72.5 g, 196 mmol, 81% yield, 95% EE, 95% pure by HPLC). |
| 81% |
|
Preparation 5 8-Benzyloxy-5-((R)-2-bromo-1-hydroxyethyl)-1H-quinolin-2-one (R)-(+)-alpha,alpha-Diphenylprolinol (30.0 g, 117 mmol) and trimethylboroxine (11.1 mL, 78 mmol) were combined in toluene (300 mL) and stirred at room temperature for 30 min. The mixture was placed in a 150 C. oil bath and liquid was distilled off. Toluene was added in 20 mL aliquots and distillation was continued for 4 h. A total of 300 mL toluene was added. The mixture was then cooled to room temperature. A 500 muL aliquot was evaporated to dryness and weighed (246 mg) to determine that the concentration of catalyst was 1.8 M. 8-Benzyloxy 5-(2-bromoacetyl)-1H-quinolin-2-one (90.0 g, 243 mmol) was placed under nitrogen and tetrahydrofuran (900 mL) was added followed by the catalyst described above (1.8 M in toluene, 15 mL, 27 mmol). The suspension was cooled to -10+-5 C. in an ice/isopropanol bath. Borane (1.0 M in THF, 294 mL, 294 mmol) was added over 4 h. The reaction was then stirred an additional 45 min at -10 C. and then methanol (250 mL) was added slowly. The mixture was concentrated under vacuum and the residue was dissolved in boiling acetonitrile (1.3 L), filtered while hot and then cooled to room temperature. The crystals were filtered, washed with acetonitrile and dried under vacuum to give the title compound (72.5 g, 196 mmol, 81% yield, 95% ee, 95% pure by HPLC). |
| 76% |
|
Preparation intermediate 4(R)-8-(Benzyloxy)-5-(2-bromo-l-hydroxyethyl)quinolin-2(l//)-one8-(Benzyloxy)-5-(2-bromoacetyl)quinolin-2(lH)-one (26.0 g, 69.9 mmol) and (R)-3,3-diphenyl-l -methyltetrahydro-3H-pyrrolo[l ,2- c][ l ,3,2]oxazaborole (21.3 g, 76.8 mmol) were azeotroped with toluene (x 3) then suspended in anhydrous THF (400 mL) under an atmosphere of nitrogen. The suspension was cooled to -20C (external temperature) and borane dimethyl sulfide complex solution (45.4 mL, 90.8 mmol, 2.0 M solution in THF) was added by syringe pump over 3 hours. After complete addition the reaction mixture was stirred for an hour before quenching with methanol (25 mL). The reaction was warmed to T over 20 minutes. The mixture was concentrated in vacuo and the residue was suspended in aqueous hydrochloric acid (500 mL, 1 M solution) and stirred at RT for 18 hours. After this time the solid was collected by filtration and washed with water (x 3). The solid was partially dissolved in ethyl acetate and heated at reflux for 2 hours. The remaining solid was removed by hot filtration and the filtrate was evaporated to afford the title compound. The solid collected from the hot ethyl acetate was again partially dissolved in ethyl acetate and heated at reflux for 2 hours then filtered to give filtrate containing pure product. This process was repeated four more times. The combined solid was recrystallised from ethyl acetate and petroleum ether to afford the title compound (20.0 g, 76%). NMR (400 MHz, DMSO): delta 10.68 (s, 1 H), 8.19 (d, J = 9.9 Hz, 1 H), 7.58 (d, J = 7.5 Hz, 2 H), 7.41 -7.36 (m, 2 H), 7.34-7.29 (m, 1 H), 7.23-7.19 (m, 2 H), 6.57 (d, J = 9.8 Hz, 1 H), 5.94 (d, J = 4.7 Hz, 1 H), 5.31 (s, 2 H), 5.25-5.19 (m, 1 H), 3.71 -3.58 (m, 2 H). |
| 76% |
|
8-(Benzyloxy)-5-(2-bromoacetyl)quinolin-2(lH)-one (26.069.9 mmol) and fR -S^-diphenyl- l-methyltetrahydro-SH-pyrrolof l ^- c][l ,3,2]oxazaborole (21.3 g, 76.8 mmol) were azeotroped with toluene (x 3) then suspended in anhydrous THF (400 mL) under an atmosphere of nitrogen. The suspension was cooled to -20C (external temperature) and borane dimethyl sulfide complex solution (45.4 mL, 90.8 mmol, 2.0 M solution in THF) was added by syringe pump over 3 hours. After complete addition the reaction mixture was stirred for one hour before quenching with methanol (25 mL). The reaction was warmed to RT over 20 minutes. The mixture was concentrated under reduced pressure and the residue was suspended in aqueous hydrochloric acid (500 mL, 1 M solution) and stirred at RT for 18 hours. After this time the solid was collected by filtration and washed with water (3 x 100 mL). The solid was partially dissolved in ethyl acetate and heated at reflux for 2 hours. The remaining solid was removed by hot filtration and the filtrate was evaporated to afford the title compound. The solid collected from the hot ethyl acetate was again partially dissolved in ethyl acetate and heated at reflux for 2 hours then filtered to give filtrate containing pure product. This process was repeated four more times. The combined solid was recrystallised from ethyl acetate and petroleum ether to afford the title compound (20.0 g, 76%). NMR (400 MHz, DMSO-d6): delta 10.68 (s, 1 H); 8.19 (d, J = 9.9 Hz, 1 H); 7.58 (d, J = 7.5 Hz, 2 H); 7.41 -7.36 (m, 2 H); 7.34-7.29 (m, 1 H); 7.23-7.19 (m, 2 H); 6.57 (d, J = 9.8 Hz, 1 H); 5.94 (d, J = 4.7 Hz, 1 H); 5.31 (s, 2 H); 5.25-5.19 (m, 1 H); 3.71 -3.58 (m, 2 H). |
| 76% |
|
8-(Benzyloxy)-5-(2-bromoacetyl)quinolin-2(1H)-one (26.0 g, 69.9 mmol) and (R)-3,3-diphenyl-1-methyltetrahydro-3H-pyrrolo[1,2-c][1,3,2]oxazaborole (21.3 g, 76.8 mmol) were azeotroped with toluene (*3) then suspended in anhydrous THF (400 mL) under an atmosphere of nitrogen. The suspension was cooled to -20 C. (external temperature), and borane dimethyl sulfide complex solution (45.4 mL, 90.8 mmol, 2.0 M solution in THF) was added by syringe pump over 3 hours. After complete addition the reaction mixture was stirred for an hour before quenching with methanol (25 mL). The reaction was warmed to RT over 20 minutes. The mixture was concentrated in vacuo and the residue was suspended in aqueous hydrochloric acid (500 mL, 1 M solution) and stirred at RT for 18 hours. After this time, the solid was collected by filtration and washed with water (*3). The solid was partially dissolved in ethyl acetate and heated at reflux for 2 hours. The remaining solid was removed by hot filtration, and the filtrate was evaporated to afford the title compound. The solid collected from the hot ethyl acetate was again partially dissolved in ethyl acetate and heated at reflux for 2 hours then filtered to give filtrate containing pure product. This process was repeated four more times. The combined solid was recrystallised from ethyl acetate and petroleum ether to afford the title compound (20.0 g, 76%). 1H NMR (400 MHz, DMSO): delta 10.68 (s, 1H), 8.19 (d, J=9.9 Hz, 1H), 7.58 (d, J=7.5 Hz, 2H), 7.41-7.36 (m, 2H), 7.34-7.29 (m, 1H), 7.23-7.19 (m, 2H), 6.57 (d, J=9.8 Hz, 1H), 5.94 (d, J=4.7 Hz, 1H), 5.31 (s, 2H), 5.25-5.19 (m, 1H), 3.71-3.58 (m, 2H). |
| 76% |
With dimethylsulfide borane complex; (3aR)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2-c][1,3,2]oxazaborole; In tetrahydrofuran; at -20℃; for 4h; |
Step 2: (R)-8-(Benzyloxy)-5-(2-bromo-1-hydroxyethyl)quinolin-2(1H)-one 8-(Benzyloxy)-5-(2-bromoacetyl)quinolin-2(1H)-one (26.0 g, 69.9 mmol) and (R)-3,3-diphenyl-1-methyltetrahydro-3H-pyrrolo[1,2-c][1,3,2]oxazaborole (21.3 g, 76.8 mmol) were azeotroped with toluene (*3) then suspended in anhydrous THF (400 mL) under an atmosphere of nitrogen. The suspension was cooled to -20 C. (external temperature) and borane dimethyl sulfide complex solution (45.4 mL, 90.8 mmol, 2.0 M solution in THF) was added by syringe pump over 3 hours. After complete addition the reaction mixture was stirred for one hour before quenching with methanol (25 mL). The reaction was warmed to RT over 20 minutes. The mixture was concentrated under reduced pressure and the residue was suspended in aqueous hydrochloric acid (500 mL, 1 M solution) and stirred at RT for 18 hours. After this time the solid was collected by filtration and washed with water (3*100 mL). The solid was partially dissolved in ethyl acetate and heated at reflux for 2 hours. The remaining solid was removed by hot filtration and the filtrate was evaporated to afford the title compound. The solid collected from the hot ethyl acetate was again partially dissolved in ethyl acetate and heated at reflux for 2 hours then filtered to give filtrate containing pure product. This process was repeated four more times. The combined solid was recrystallised from ethyl acetate and petroleum ether to afford the title compound (20.0 g, 76%). 1H NMR (400 MHz, DMSO-d6): delta 10.68 (s, 1H); 8.19 (d, J=9.9 Hz, 1H); 7.58 (d, J=7.5 Hz, 2H); 7.41-7.36 (m, 2H); 7.34-7.29 (m, 1H); 7.23-7.19 (m, 2H); 6.57 (d, J=9.8 Hz, 1H); 5.94 (d, J=4.7 Hz, 1H); 5.31 (s, 2H); 5.25-5.19 (m, 1H); 3.71-3.58 (m, 2H). |
| 76% |
With dimethylsulfide borane complex; (3aR)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2-c][1,3,2]oxazaborole; In tetrahydrofuran; toluene; at -20 - 20℃;Inert atmosphere; |
8-(Benzyloxy)-5-(2-bromoacetyl)quinolin-2(lH)-one (26.0 g, 69.9 mmol) and (^-3,3-diphenyl- 1 -methyltetrahydro-3H-pyrrolo[ 1 ,2-c][ 1 ,3,2]oxazaborole (21.3 g, 76.8 mmol) were azeotroped with toluene (x 3) then suspended in anhydrous THF (400 mL) under an atmosphere of nitrogen. The suspension was cooled to -20 C (external temperature) and borane dimethyl sulfide (BH3-Me2S) complex solution (45.4 mL, 90.8 mmol, 2.0 M solution in THF) was added by syringe pump over 3 hours. After complete addition the reaction mixture was stirred for one hour before quenching with methanol (25 mL). The reaction was warmed to RT over 20 minutes. The mixture was concentrated under reduced pressure and the residue was suspended in aqueous hydrochloric acid (500 mL, 1 M solution) and stirred at RT for 18 hours. After this time the solid was collected by filtration and washed with water (3 x 100 mL). The solid was partially dissolved in ethyl acetate and refluxed for 2 hours. The remaining solid was removed by hot filtration and the filtrate was evaporated to afford the title compound. The solid collected from the hot ethyl acetate was again partially dissolved in ethyl acetate and refluxed for 2 hours then filtered to give filtrate containing pure product. This process was repeated four more times. The combined solid was recrystallised from ethyl acetate and petroleum ether to afford the title compound (20.0 g, 76%). 1HNMR (400 MHz, DMSO-d6): delta 10.68 (s, 1 H); 8.19 (d, J = 9.9 Hz, 1 H); 7.58 (d, J = 7.5 Hz, 2 H); 7.41-7.36 (m, 2 H); 7.34-7.29 (m, 1 H); 7.23-7.19 (m, 2 H); 6.57 (d, J = 9.8 Hz, 1 H); 5.94 (d, J = 4.7 Hz, 1 H); 5.31 (s, 2 H); 5.25-5.19 (m, 1 H); 3.71-3.58 (m, 2 H). |
| 76% |
|
8-(l3enzyloxy)-5-(2-bromoacetyl)quinolin-2(1 H)- one (26.0 g, 69.9 mmol) and (R)-3,3-diphenyl-1-methyltet- rahydro-3H-pyrrolo[1 ,2-c] [1 ,3,2]oxazaborole (21.3 g, 76.8 mmol) were azeotroped with toluene (x3) then suspended in anhydrous THF (400 mE) under an atmosphere of nitrogen. The suspension was cooled to -20 C. (external temperature) and borane dimethyl sulfide complex solution (45.4 mE, 90.8 mmol, 2.0 M solution in THF) was added by syringe pump over 3 hours. Afier complete addition the reaction mixture was stirred for one hour before quenching with methanol (25 mE). The reaction was warmed to room temperature over 20 minutes. The mixture was concentrated under reduced pressure and the residue was suspended in aqueous hydrochloric acid (500 mE, 1 M solution) and stirred at room temperature for 18 hours. After this time the solid was collected by filtration and washed with water (3x100 mE). The solid was partially dissolved in ethyl acetate and heated at reflux for 2 hours. The remaining solid was removed by hot filtration and the filtrate was evaporated to afford the title compound. The solid collected from the hot ethyl acetate was again partially dissolved in ethyl acetate and heated at reflux for 2 hours then filtered to give filtrate containing pure product. This process was repeated four more times. The combined solid was recrystallised from ethyl acetate and petroleum ether to afford the title compound (20.0 g, 76%). 1H NMR (400 MHz, DMSO-d5): oe 10.68 (s, 1H),8.19 (d, J=9.9 Hz, 1H), 7.58 (d, J=7.5 Hz, 2H), 7.41-7.36 (m,2H), 7.34-7.29 (m, 1H), 7.23-7.19 (m, 2H), 6.57 (d, J=9.8 Hz,1H), 5.94 (d, J=4.7 Hz, 1H), 5.31 (s, 2H); 5.25-5.19 (m, 1H),3.71-3.58 (m, 2H). |
| 76% |
With borane dimethyl sulfide complex; (3aR)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2-c][1,3,2]oxazaborole; In tetrahydrofuran; toluene; at -20℃; for 4h;Inert atmosphere; |
8-(benzyloxy)-5-(2-bromoacetyl)quinolin-2(1H)-one (26.0 g, 69.9 mmol) and (R)- 3 ,3-diphenyl- 1 -methyltetrahydro-3H-pyrrolo [ 1 ,2-c] [ 1 ,3 ,2]oxazaborole (21.3 g, 76.8 mmol) were azeotroped with toluene (x 3) then suspended in anhydrous THF (400 mL) under an atmosphere of nitrogen. The suspension was cooled to -20C (external temperature) and borane dimethyl sulfide complex solution (45.4 mL, 90.8 mmol, 2.0 M solution in THF) was added by syringe pump over 3 hours. After complete addition the reaction mixture was stirred for one hour before quenching with methanol (25 mL). The reaction was warmed to room temperature over 20 minutes. The mixture was concentrated under reduced pressure and the residue was suspended in aqueous hydrochloric acid (500 mL, 1 M solution) and stirred at room temperature for 18 hours. After this time the solid was collected by filtration and washed with water (3 x 100 mL). The solid was partially dissolved in ethyl acetate and heated at reflux for 2 hours. The remaining solid was removed by hot filtration and the filtrate was evaporated to afford the title compound. The solid collected from the hot ethyl acetate was again partially dissolved in ethyl acetate and heated at reflux for 2 hours then filtered to give filtrate containing pure product. This process was repeated four more times. The combined solid was recrystallised from ethyl acetate and petroleum ether to afford the title compound (20.0 g, 76%). NMR (400 MHz, DMSO-d6): delta 10.68 (s, 1 H), 8.19 (d, J= 9.9 Hz, 1 H), 7.58 (d, J = 7.5 Hz, 2 H), 7.41-7.36 (m, 2 H), 7.34-7.29 (m, 1 H), 7.23-7.19 (m, 2 H), 6.57 (d, J = 9.8 Hz, 1 H), 5.94 (d, J = 4.7 Hz, 1 H), 5.31 (s, 2 H); 5.25-5.19 (m, 1 H), 3.71-3.58 (m, 2 H). |
| 76% |
With dimethylsulfide borane complex; (3aR)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2-c][1,3,2]oxazaborole; In tetrahydrofuran; toluene; at -20℃; for 4h;Inert atmosphere; |
8-(Benzyloxy)-5-(2-bromoacetyl)quinolin-2(lH)-one (26.0 g, 69.9 mmol) and (R)- 3,3-diphenyl-l-methyltetrahydro-3H-pyrrolo[l,2-c][l,3,2]oxazaborole (21.3 g, 76.8 mmol) were azeotroped with toluene (x 3) then suspended in anhydrous THF (400 mL) under an atmosphere of nitrogen. The suspension was cooled to -20C (external temperature) and borane dimethyl sulfide complex solution (45.4 mL, 90.8 mmol, 2.0 M solution in THF) was added by syringe pump over 3 hours. After complete addition the reaction mixture was stirred for one hour before quenching with methanol (25 mL). The reaction was warmed to room temperature over 20 minutes. The mixture was concentrated under reduced pressure and the residue was suspended in aqueous hydrochloric acid (500 mL, 1 M solution) and stirred at room temperature for 18 hours. After this time the solid was collected by filtration and washed with water (3 chi 100 mL). The solid was partially dissolved in ethyl acetate and heated at reflux for 2 hours. The remaining solid was removed by hot filtration and the filtrate was evaporated to afford the title compound. The solid collected from the hot ethyl acetate was again partially dissolved in ethyl acetate and heated at reflux for 2 hours then filtered to give filtrate containing pure product. This process was repeated four more times. The combined solid was recrystallised from ethyl acetate and petroleum ether to afford the title compound (20.0 g, 76%). NMR (400 MHz, DMSO-d6): delta 10.68 (s, 1 H), 8.19 (d, J= 9.9 Hz, 1 H), 7.58 (d, J= 7.5 Hz, 2 H), 7.41-7.36 (m, 2 H), 7.34-7.29 (m, 1 H), 7.23-7.19 (m, 2 H), 6.57 (d, J= 9.8 Hz, 1 H), 5.94 (d, J= 4.7 Hz, 1 H), 5.31 (s, 2 H); 5.25-5.19 (m, 1 H), 3.71-3.58 (m, 2 H). |
|
|
A mixture of trimethylboroxine (0.74ml 5.34mmol) and (R)-2-(diphenylhydroxymethyl)pyrrolidine (2.03g, 8.0mmol) in toluene (13.6ml) was stirred for 40 minutes. Toluene (13.6ml) was added and the mixture was concentrated to 8 ml by distillation at 1 atm. Another portion of toluene (13.6 ml) was added and the mixture was concentrated to 8 ml by distillation at 1 atm. This procedure was repeated another three times using anhydrous toluene. A suspension of the compound from preparation 1 (12.0g, 32.2mmol) in anhydrous tetrahydrofuran (180ml) under nitrogen was cooled to -5C using an ice-isopropanol bath. The toluene solution of the oxoborolidine catalyst prepared above was added in one portion via syringe. Borane dimethyl sulphide complex (5.0 ml, 50mmol) was dissolved in anhydrous tetrahydrofuran (50ml) under nitrogen. A portion of this (40ml, 40mmol) was added at -6C via syringe using a syringe pump over 3.5 hours. Methanol (40ml) was added slowly keeping the reaction mixture between -5C and 0C. The mixture was stirred for 30 minutes and allowed to warm to room temperature. The solvent was removed under reduced pressure and the residue was slurried in methanol (200ml) and concentrated to dryness (twice). The crude product was dissolved in dichloromethane (1300ml) containing a little methanol, and washed with hydrochloric acid (1M, 240ml) and water (2 × 300ml). The organic solution was dried (MgSO4) and concentrated under reduced pressure and the product recrystallised from glacial acetic acid to give the desired product as an off-white solid, 11.0g.1H nmr (CDCl3, 400MHz) delta: 2.88 (brd, 1H,), 3.58 (dd, 1H), 3.62 (dd, 1H), 5.16 (s, 2H), 5.29 (m, 1H), 6.68 (d, 1H), 7.01 (d, 1H), 7.25 (d, 1H), 7.39 (s, 5H), 8.04 (d, 1H), 9.18 (br s, 1H). LRMS :m/z ES+ 374, 376 [MH+] |
|
With borane-THF;Trimethylboroxine; (R)-alpha,alpha-diphenylprolinol; In tetrahydrofuran; toluene; at -10℃; for 4.75h; |
(R)-(+)-alpha,alpha-Diphenylprolinol (30.0 g, 117 mmol) and trimethylboroxine (11.1 mL, 78 mmol) were combined in toluene (300 mL) and stirred at room temperature for 30 min. The mixture was placed in a 150 C. oil bath and liquid was distilled off. Toluene was added in 20 mL aliquots and distillation was continued for 4 h. A total of 300 mL toluene was added. The mixture was then cooled to room temperature. A 500 muL aliquot was evaporated to dryness and weighed (246 mg) to determine that the concentration of catalyst was 1.8 M. 8-Benzyloxy 5-(2-bromoacetyl)-1H-quinolin-2-one (90.0 g, 243 mmol) was placed under nitrogen and tetrahydrofuran (900 mL) was added followed by the catalyst described above (1.8 M in toluene, 15 muL, 27 mmol). The suspension was cooled to -10+/-5 C. in an ice/isopropanol bath. Borane (1.0 M in THF, 294 mL, 294 mmol) was added over 4 h. The reaction was then stirred an additional 45 min at -10 C. and then methanol (250 mL) was added slowly. The mixture was concentrated under vacuum and the residue was dissolved in boiling acetonitrile (1.3 L), filtered while hot and then cooled to room temperature. The crystals were filtered, washed with acetonitrile and dried under vacuum to give the title compound (72.5 g, 196 mmol, 81% yield, 95% ee, 95% pure by HPLC). |
|
With borane;(3aR)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2-c][1,3,2]oxazaborole; In tetrahydrofuran; at -5℃; for 3.75h; |
Using a procedure described in Mathre et al., J. Org. Chem., 1991, 56, 751-762, a catalyst was prepared as follows. (R)-(+)-alpha,alpha-Diphenylprolinol (10.0 g, 39 mmol) and trimethylboroxine (3.7 mL, 26 mmol) were combined in toluene (200 mL) and stirred at room temperature for 30 min. The mixture was placed in a 150 C. oil bath and 150 mL liquid was distilled away. Toluene (50 mtL) was added, and another 50 mL of distillate was collected. Another portion of toluene (50 mL) was added and a further 50 mL of distillate was collected. A 1.00 mL aliquot of the material remaining in the pot was evaporated to dryness and weighed (241.5 mg) to determine that the concentration of catalyst was 0.87 M. 5-(2-Bromo-1-oxy)ethyl-8-benzyloxy-2(1H)-quinolinone (R) (30.0 g, 81 mmol) was suspended in, tetrahydrofuran (1.2 L) under a nitrogen atmosphere and the catalyst from above (13 mL, 11 mmol) was added. The suspension was cooled to -5 C. in an ice/isopropanol bath and borane (1.0 M in THF, 97 mL, 97 mmol) was added over 3 h. The reaction was stirred an additional 45 min at -5 C., then methanol (200 mL) was added slowly. The mixture was concentrated under vacuum to give 5-(2-bromo-(R)-1-hydroxy)ethyl-8-benzyloxy-2(1H)-quinolinone (FF). |
|
|
Under an argon flow, 8-benzyloxy-5-(2-bromoacetyl)-1H-quinolin-2-one (374 g, 1.00 mol) was suspended in dehydrated tetrahydrofuran (3.8 L), CBS catalyst (27.8 g) was added thereto, and the mixture was stirred at -55 to -45 C. for 40 minutes. After adding dropwise a 0.9 M tetrahydrofuran solution of borane-tetrahydrofuran complex (1.27 L) at the same temperature, the reaction mixture was gradually warmed to 0 C. After adding methanol (1.3 L) dropwise, insoluble matter was removed by filtration and washed with tetrahydrofuran. The filtrate and the washing were mixed and concentrated under reduced pressure. To the residue was added purified water (6.3 L), and the mixture was stirred at room temperature overnight. The precipitate was collected by filtration and washed with purified water. Further, the precipitate was suspension-washed with ethyl acetate (4.8 L), collected by filtration, and dried under reduced pressure to obtain 8-benzyloxy-5-((R)-2-bromo-1-hyroxyethyl)-1H-quinolin-2-one (278 g). 1H-NMR(400 MHz, CDCl3) 5 9.26(s, 1H), 8.08(d, J=10.0 Hz, 1H), 7.37-7.45(m, 5H), 7.28(d, J=8.0 Hz, 1H), 7.05(d, J=8.0 Hz, 1H), 6.72(d, J=10.0 Hz, 1H), 5,33 (dd, J=9.0 Hz, 4.0 Hz, 1H), 5.19(s, 2H), 3.71-3.74(m, 2H) |
|
|
ii) 8-(Benzyloxy)-5-[(lR)-2-bromo-l-hydroxyethyl]quinolin-2(liJ)-one; The product of step (i) (8-(benzyloxy)-5-(bromoacetyl)quinolin-2(lH)-one) (5.0 g) was placed in an oven-dried flask and dried for 2 d under vacuum at 4O0C, then to it was added <n="65"/>dry THF (100 mL). (R)-(+)-2-Methyl-CBS-oxazaborolidine (2.20 mL, l.OM in toluene) was added and the mixture cooled to -1O0C. Borane-THF complex (16.2 mL, 1.0M) was added over 3.5h using a syringe pump. The reaction mixture was stirred at -10 to O0C for Ih. Methanol (100 mL) was added and the volatiles removed in vacuo, followed by azeotroping with methanol (x3). The residue was dissolved in boiling acetonitrile (140 mL), allowed to cool slowly and filtered to afford the sub-title compound as a pale yellow solid. Yield: 3.8 g MS APCI+ 374/376[M+H]+ |
|
|
Example 6 Preparation of 8-benzyloxy-5-[(R)-(2-bromo-1-hydroxyethyl)]-carbostyril (compound (Ig); R1 = benzyl)A dry 5 liter flask equipped with a mechanical stirrer, thermometer, addition funnel and refluxing condenser was charged with 5-Bromoacetyl-8-benzyloxycarbostyril (100 gms/ 0.268 moles) and dry THF (1.2 liter) under argon. A solution of (R)-tetrahydro-i-methyl- 3,3-diphenyl-(1 H,3H)-pyrrolo[1 ,2-c][1 ,3,2]-oxazoborolidine catalyst in toluene (43 ml/ 0.388 moles) was added and reaction mass was chilled to 0-20C. Then 1M solution of borane- methyl sulfide (32.42 ml/ 0.3417 moles) in THF (342ml) was added in 3 hours while maintaining temperature at 0-2C. The reaction was stirred for another 15-20 minutes at same temperature. The reaction mass was quenched by addition of methanol (171 ml) in 15-20 minutes . The temperature of resulting solution was raised to 25-300C and concentrated to a volumes of 500 ml under vacuum at 50C. To this concentrate was added a mixture of water (1.45 liters) and concentrated HCI (74 ml) in 10-15 minutes. The resulting suspension was stirred for 30 minutes at 25C. The solid 8-benzyloxy- 5-[( R )-(2- bromo-1-hydroxyethyl)]- carbostyril obtained was isolated by filtration and washed with water till neutral pH, dried under vacuum at 60-65 C for 12-14 hours. Yield- 70-73 gms |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping