* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
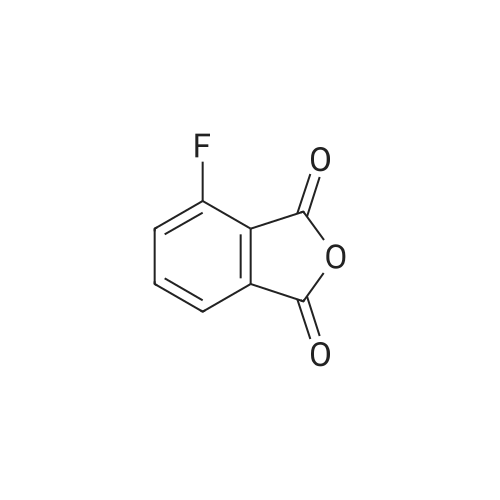
With thionyl chloride; potassium fluoride; urea; In sulfolane; water; acetone;
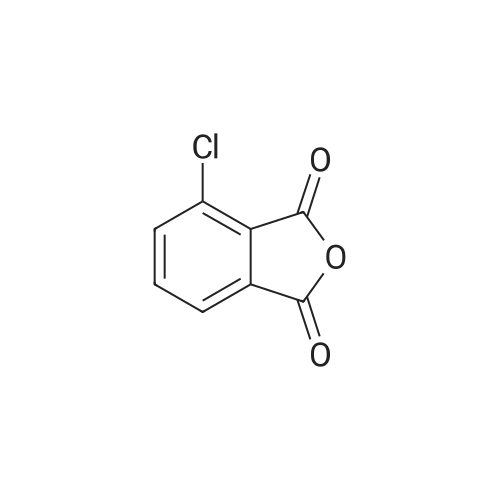
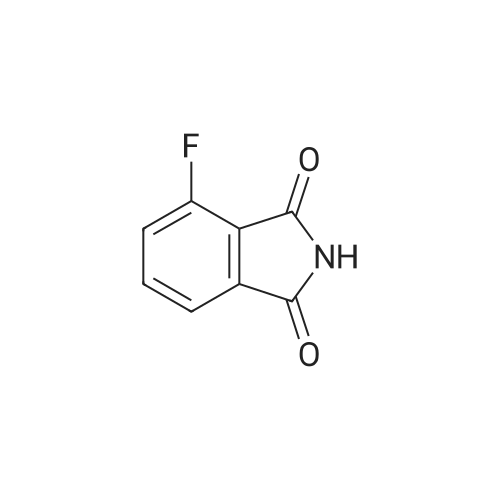
EXAMPLE 2 500 parts (2.739 moles) of <strong>[117-21-5]3-chlorophthalic anhydride</strong> and 49.9 parts of thionyl chloride are added to 820 parts of sulfolane, while stirring. The mixture is stirred for 30 minutes at 100 C., after which the evolution of gas is complete. Excess thionyl chloride is stripped off under reduced pressure from a water pump, after which 175 parts of potassium fluoride are added and stirring is continued for 6 hours at 210 C. The mixture is cooled to 60 C., and 82 parts (1.37 moles) of urea are then added. The mixture is stirred for 1 hour 30 minutes at 130 C., when evolution of gas is complete, after which stirring is continued for a further 15 minutes at 170 C. The mixture is then cooled to room temperature, 300 parts of acetone are added, while stirring, and the predominantly inorganic precipitate is filtered off under suction. The filtrate is freed from acetone in a rotary evaporator, and the residue is then distilled over a 10 cm packed column at a bath temperature of 140-170 C. under 0.4 mbar. The distillation residue is stirred with water, and the product is filtered off under suction, washed with methyl tert.-butyl ether and dried, 382 parts (84.5% of theory) of 3-fluorophthalimide of melting point 176-179 C. being obtained. By treating the inorganic residue with 300 parts of acetone, separating off the extract and evaporating it down under reduced pressure, a further 29 parts (6.8% of theory) of 3-fluorophthalimide of melting point 166-178 C. are isolated.
With formamide; In neat (no solvent); for 0.333333h;Milling; Heating; Green chemistry;
General procedure: A mixture of anhydride (1 mmol), formamide (1.1 mmol for monoanhydrides and 2.2 mmol for dianhydrides) and 1 g clay was ground together in a mortar using pestle for the time described in Table 1. The reaction mixture was warmed. After completing the reaction (monitored by TLC, after observing no anhydride presence in the reaction mixture), the product was extracted by washing clay with chloroform (2×15 mL), the solvent was removed under vacuum to afford the relevant N-unsubstituted cyclic imide. The solid imide was washed thoroughly with water, dried, and then recrystallized from ethanol. The solid clay portion was washed with methanol and dried at 120 C under a reduced pressure to be reused in the subsequent reactions which showed the gradual decrease in the activity (Table 1). Isolated products were characterized by melting points, IR, 1H NMR spectrometric data and were compared with the literature or authentic samples.
83%
With formamide; at 130℃; for 1h;Inert atmosphere;
Under argon, 10.0 g (60.2 mmol) of 4-fluoro-2-benzofuran-1,3-dione were stirred at 130 C. in 50 ml of formamide for 1 h. The cooled reaction mixture was then added to ice-water. A solid precipitated out. This solid was filtered off with suction and washed with water. The product was dried under high vacuum. This gave 8.3 g (83% of theory) of the target compound.LC-MS (method 11): Rt=0.57 min; m/z=166 (M+H)+.1H-NMR (400 MHz, DMSO-d6): δ=11.47 (br. s, 1H), 7.87 (td, 1H), 7.69-7.61 (m, 2H).
73%
With formamide; at 125℃; for 1 - 5h;
The best result occurs when the starting material is run 0.5 M in formamide and heated to 125 C for 1 to 5 h depending on scale. Starting material is not soluble in fonnamide until the temperature is > 60 C. Upon completion of reaction as monitored by LC/MS (apcineg), the heat is removed and 3 times the volume of the reaction of water is added. Next, the reaction is allowed to warm to room temperature and stirred until a pale yellow precipitate has formed. The yellow solid product is filtered off and washed with water before drying overnight to give yields between 70-77 %.
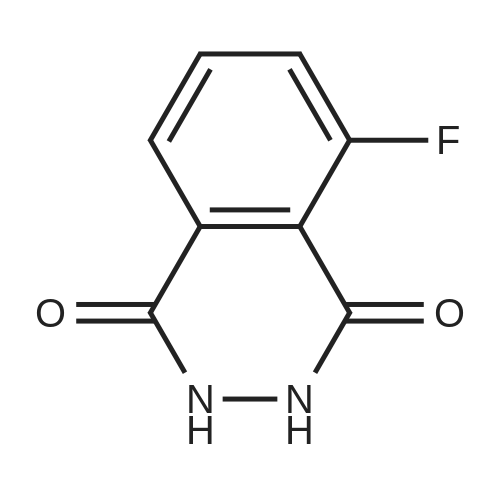
With ammonia; In water; at 280℃; for 0.5h;
In a 50 ml flask was placed 3-Fluorophthalic anhydride (1.0 g, 6 mmol) and aqueous NH3 (1.6 g, 24 mmol). The mixture was heated to 280 0C within 30 minutes and then the flask was cooled to room temperature. 0.93 g of compound 13 were isolated as a yellow solid. LC-MS (ESI, positive): 166 [M+H]+.
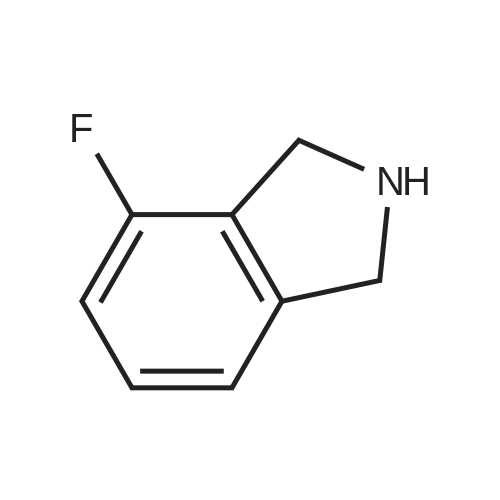
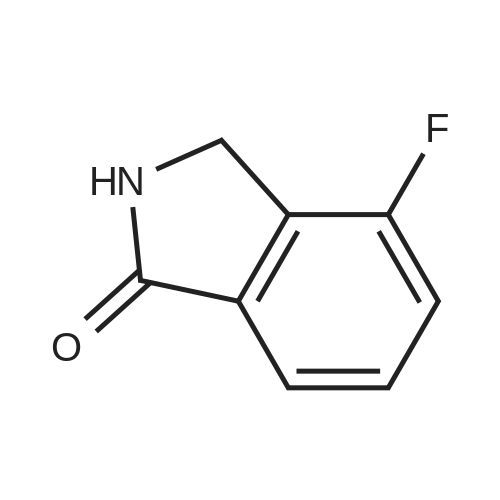
To the starting material in a round bottom flask was added 4 equivalents of 1 M BH3- THF drop wise using an addition funnel to form a golden solution which upon heating and stirring turned copper in color. The reaction was then heated at reflux for 18 h. [00590] The reaction is then cooled to room temperature (rt) and then to 0 C in an ice bath. 4 equivalents of MeOH are added drop wise and the ice bath removed so quenched reaction can warm to rt. Reaction color turns dark during this warming process. Next, 6 N HC1 was added drop wise at rt until pH paper showed reaction to be acidic and the reaction refluxed (63 C) for 1 h. The reaction was then cooled to rt. At this point the reaction was concentrated and washed with Et2O (2x) and DCM (2x). The aqueous layer was then brought to pH = 11 with NaOH pellets. More water was added and the aqueous layer was extracted with ether (4x). The combined extracts were dried over Na2S04 and concentrated to give a light tan colored oil product, which was used directly. Mass recovery is always slightly higher than theoretical, but material is used crude like this to give > 80 % yield in the next step.
To compound 13 (4.0 g, 24.2 mmol) in a round bottom flask was added 1M BH3 in THF solution (97 mL, 97 mmol) dropwise at room temperature. The resulting solution was warmed to reflux for 18 hours. Then the reaction mixture was cooled to 0 0C and methanol (3.1 g, 97 mmol) was added dropwise. The resulting mixture was warmed up to room temperature and then 6 M HCl was added dropwise to adjust the reaction pH to 3, followed by refluxed for 1 hour. <n="54"/>After the reaction was completed, the solvents were removed under reduced pressure to give an brown oil. The residue was washed with Et2O (2x50 ml) and CH2Cl2 (2x50 mL). The aqueous phase was adjusted to pH 11 with NaOH. Then the aqueous layer was extracted with ether (4x50 mL), dried over Na2SO4, and filtered. Solvents were removed under reduced pressure to give a dark red residue. The pure compound was purified by distillation (2 mmHg, 45 0C) to give compound 14 (1.2 g).
With sodium tetrahydroborate; ethanol; at 0 - 20℃;
7458 mg (12.1 mmol) of sodium borohydride were added a little at a time to a suspension, cooled to 0 C., of 4.0 g (24.2 mmol) of 4-fluoro-1H-isoindole-1,3(2H)-dione in 40 ml of ethanol. After the addition had ended, cooling was removed and the mixture was warmed to RT. After 20 min of stirring, a further 458 mg (12.1 mmol) of sodium borohydride were added at RT. After a further 30 min at RT, 1 N hydrochloric acid was added carefully to the reaction mixture and the pH was adjusted to neutral. The mixture was concentrated under reduced pressure and the residue was taken up in dichloromethane and filtered through Celite. The collected filtrate was concentrated under reduced pressure. This gave 4.05 g of 4-fluoro-3-hydroxy-2,3-dihydro-1H-isoindol-1-one, which was directly reacted further as crude product.4.05 g (about 24.2 mmol) of 4-fluoro-3-hydroxy-2,3-dihydro-1H-isoindol-1-one were initially charged in 10 ml of dichloromethane, 22.4 ml (290.8 mmol) of trifluoroacetic acid and 7.7 ml (48.5 mmol) of triethylsilane were added and the mixture was stirred at RT overnight. The reaction mixture was then concentrated and the residue was dissolved in dichloromethane and washed with sodium bicarbonate solution. The crude product was purified by chromatography on silica gel (mobile phase cyclohexane/ethyl acetate 3:1 to 1:1). This gave 890 mg (24.2% of theory) of the target compound.LC-MS (method 12): Rt=1.18 min; m/z=152 (M+H)+.1H-NMR (400 MHz, DMSO-d6): δ=8.77 (br. s, 1H), 7.58-7.51 (m, 2H), 7.49-7.40 (m, 1H), 4.46 (s, 2H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping