* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
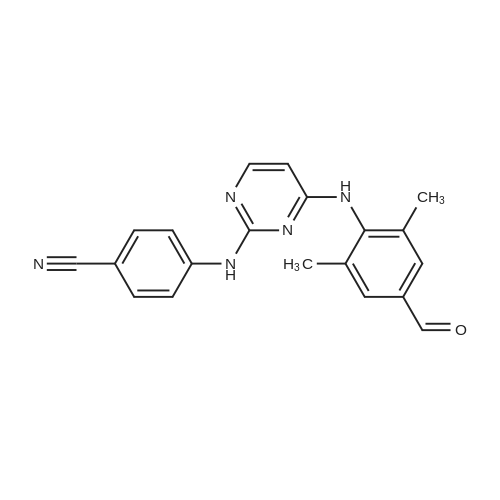
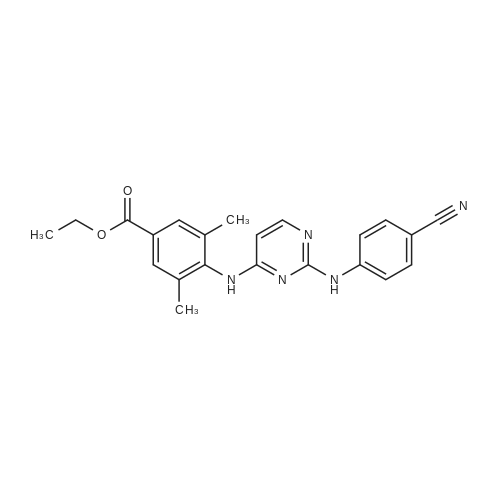
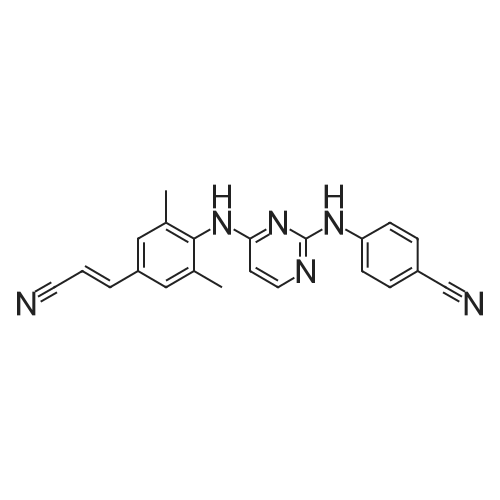
4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile[ No CAS ]
[ 500287-72-9 ]
Yield
Reaction Conditions
Operation in experiment
5%; 7%
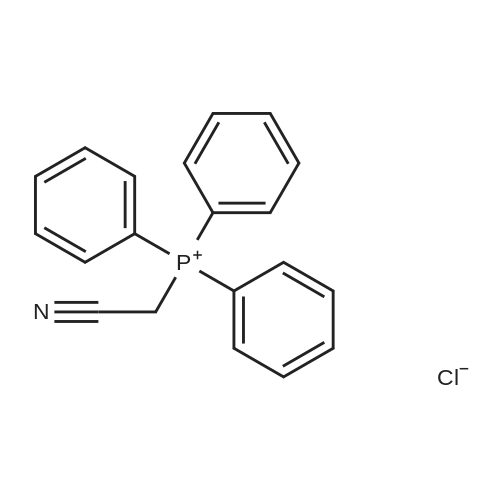
A mixture of (cyanomethyl)triphenylphosphonium chloride (0.0022 mol) and potassium tert.-butoxide (0.0022 mol) in THF (7 ml) was stirred at 5 C. for 30 minutes under N2 flow, then stirred at 5 C. for 30 minutes. A mixture of intermediate 13 (0.0015 mol) in THF (7 ml) was added. The mixture was stirred for 8 hours in darkness, poured out into H2O and extracted with CH2Cl2. The organic layer was separated, dried (MgSO4), filtered and the solvent was evaporated. The residue (1.4 g) was purified by column chromatography over silica gel (eluent: toluene/iPrOH/NH4OH 96/4/0.1; 15-40 mum). Two fractions (F1, F2) were collected and the solvent was evaporated. Yield: 0.165 g of F1 (E/Z=32/68) (30%) and 0.225 g of F2 (E/Z=0/10) (41%). F2 was crystallized from CH3CN/diethyl ether. Yield: 0.036 g of compound 1 (7%). F1 was purified by column chromatography over kromasyl (eluent: toluene/iPrOH 98/2; 5 mum). The pure fractions were collected and the solvent was evaporated. Yield: 0.029 g of compound 10 (5%).
4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile[ No CAS ]
[ 500287-72-9 ]
Yield
Reaction Conditions
Operation in experiment
55%
With triethylamine; tris-(o-tolyl)phosphine;palladium diacetate; In acetonitrile; at 150℃;
A mixture of intermediate (IV-a) (0.00021 mol), prepared according to Example A4, acrylonitrile (CH2=CH-CN) (0.00213 mol), Pd (OAc) 2 (0.000043 mol), N,N-diethylethanamine (0.000043 mol) and tris (2-methylphenyl) phosphine (0.00021 mol) in CH3CN (7 ml) was stirred in a sealed vessel at 150C overnight. H20 was added. The mixture was extracted with CH2C12. The organic layer was separated, dried (MgS04), filtered and the solvent was evaporated. The residue (0.15 g) was purified by column chromatography over silica gel (eluent: CH2Cl2/ethyl acetate 80/20; 15-40 mum). Fraction 1 was collected and the solvent was evaporated, yielding 0.045g of 4-[[4-[[4- [(2-cyanoethenyl)-2,6-dimethylphenyl] amino]-2-pyrimidinyl]amino] benzonitrile (E/Z=80/20). The solid was crystallized from diethylether. Yield: 0.035g of 4-[[4-[[4- (2-cyanoethenyl)-2, 6-dimethylphenyl] amino]-2-pyrimidinyl] amino] benzonitrile (E) (Compound X) [(55%).]
With palladium diacetate; triethylamine; tris-(o-tolyl)phosphine; In acetonitrile; at 150℃;Sealed tube;
A mixture of intermediate 58 (0.00021 mol), prepared according to Example A11, acrylonitrile (CH2?CH-CN) (0.00213 mol), Pd(OAc)2 (0.000043 mol), N,N-diethylethanamine (0.000043 mol) and tris(2-methylphenyl)phosphine (0.00021 mol) in CH3CN (7 ml) was stirred in a sealed vessel at 150 C. overnight H2O was added. The mixture was extracted with CH2Cl2. The organic layer was separated, dried (MgSO4), filtered and the solvent was evaporated. The residue (0.15 g) was purified by column chromatography over silica gel (eluent: CH2Cl2/ethyl acetate 80/20; 15-40 mum). Fraction 1 was collected and the solvent was evaporated, yielding 0.045 g of 4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (E/Z=80/20). The solid was crystallized from diethylether. Yield: 0.035 g of 4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (E) (compound 1) (55%).
4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile[ No CAS ]
[ 500287-72-9 ]
Yield
Reaction Conditions
Operation in experiment
With sodium acetate;palladium 10% on activated carbon; In N,N-dimethyl acetamide; at 140℃;
4, 41g (10 mmol) of intermediate (IV-b) and 15 ml of N,N-dimethylacetamide were brought in a 100 ml flask under nitrogen. To this mixture were added 0,98g of sodium acetate (12 mmol), 107 mg (0,1 mmol Pd) of Pd/C] 10% (wet) and [1] mi (15 mmol) of acrylonitrile. The mixture was heated at 140C and the evolution of the reaction was followed by liquid chromatography. The reaction yielded 4-[[4-[[4-(2-cyanoethenyl)-] 2,6-dimethylphenyl] amino]-2-pyrimidinyl] amino] benzonitrile (E/Z=80/20) which can be worked up to yield 4-[[4-[[4-(2-cyanoethenyl)-2, 6-dimethylphenyl] amino]-2- pyrimidinyl] amino] benzonitrile (E) as described above in Example B2.
With palladium 10% on activated carbon; sodium acetate; In N,N-dimethyl acetamide; at 140℃;Inert atmosphere;
4.41 g (10 mmol) of intermediate 59 and 15 ml of N,N-dimethylacetamide were brought in a 100 ml flask under nitrogen. To this mixture were added 0.98 g of sodium acetate (12 mmol), 107 mg (0.1 mmol Pd) of Pd/C 10% (wet) and 1 ml (15 mmol) of acrylonitrile. The mixture was heated at 140 C. and the evolution of the reaction was followed by liquid chromatography. The reaction yielded 4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (E/Z=80/20) which can be converted to 4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (E) as described above in Example B1Ba).
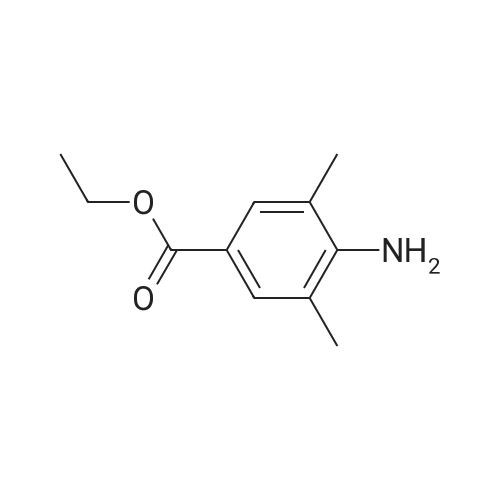
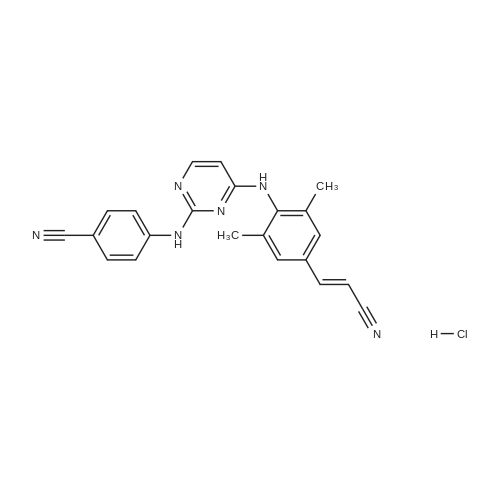
(2E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile hydrochloride[ No CAS ]
[ 244768-32-9 ]
[ 500287-72-9 ]
Yield
Reaction Conditions
Operation in experiment
89.6%
b) A mixture of 93.9 g (0. 45 mol) of the hydrochloric acid salt of intermediate [(II),] prepared according to Example A2, and 103.8 g (0.45 mol) of intermediate (III-a) in 0.9 1 of acetonitrile was prepared under nitrogen atmosphere. The mixture was stirred and refluxed for 24 hours, then allowed to cool to 50 C. A solution of K2C03 (124.4 g, 0.9 mol) in H20 (0.45 1) was added over a period of 15-20 minutes at 40-50 C, followed by stirring for 1 hour at 50 C. The precipitate was separated and washed twice with 0.045 1 of acetonitrile, followed by drying at 50C under reduced pressure. 73.3 g of the obtained solid and 400 ml of EtOH were mixed and refluxed for 2 hours, then allowed to cool to room temperature. The precipitate was filtered and the residue was washed with 50 ml of EtOH. The obtained residue was dried overnight at 50C under reduced pressure. Yield: 65.7 g (89. 6 %) of 4-[[4-[[4-(2-cyanoethenyl)-2, 6- dimethylphenyl] amino]-2-pyrimidinyl] amino] benzonitrile (E) (Compound X).
68.6%
A mixture of 93.9 g (0.45 mol) of the hydrochloric acid salt of intermediate (II), prepared according to Example A2, and 109 g (0.4725 mol) of intermediate (III-a) in 1.81 of of acetonitrile was prepared under nitrogen atmosphere. The mixture was stirred and refluxed for 69 hours, then allowed to cool to 55 C. The mixture was filtered and the residue was washed with 200 ml of acetonitrile, followed by drying under reduced pressure at 50C overnight. 144, 6 g (0.3666 mol) of the obtained solid was brought in 1 l of K2CO3 10% aqueous solution. The mixture was stirred at room temperature followed by filtration. The obtained residue was washed twice with water followed by drying at 50C under reduced pressure. The residue was brought in 6.55 1 isopropanol and the mixture was refluxed, then stirred overnight and filtered at room temperature. The residue was dried at 50C under reduced pressure. Yield: 113.2 g [(68.] 6 %) of 4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino-2-pyrimidinyl]amino]benzonitrile (E) (Compound X).
With toluene-4-sulfonic acid; In 1,4-dioxane; at 100 - 110℃; for 14h;Product distribution / selectivity;
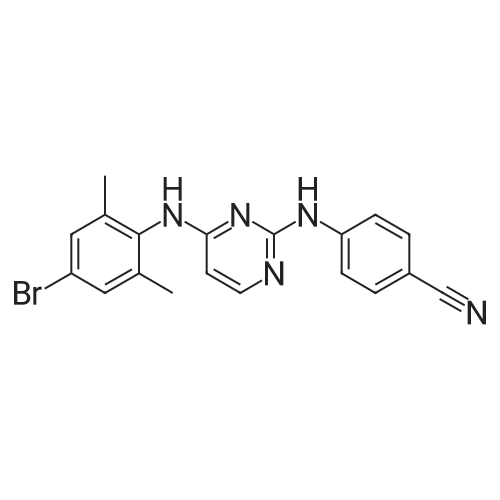
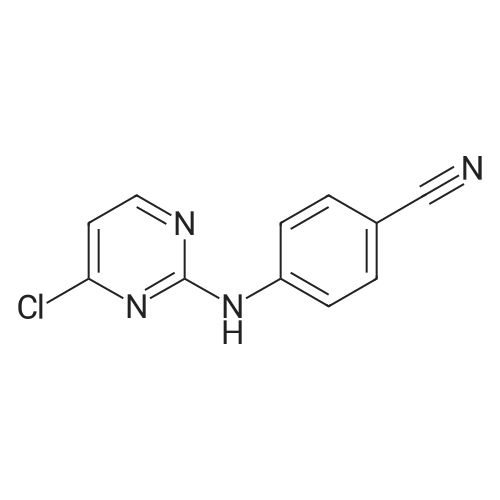
Example 8:Preparation of RilpivirineTo a mixture of 4-(4-chloropyrimidin-2-ylamino)benzonitrile (4.5 gm) as obtained in example 3, (E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile hydrochloride (4.07 gm) as obtained in example 7 and p-toluenesulfonic acid monohydrate (4.45 gm) was added 1,4-dioxane (90 ml) under stirring. The mixture was then heated to 100 to 110C and stirred for 14 hours. The solution was then cooled to room temperature and then added saturated sodium bicarbonate solution. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers were dried with sodium sulfate and then concentrated to obtain a crude solid.The crude solid obtained above was dissolved in acetone and stirred for 1 hour at room temperature. The separated solid was filtered and then dried to obtain 4 gm of rilpivirine.
135 g
In 1-methyl-pyrrolidin-2-one; at 85 - 95℃; for 16h;
To a mixture of 4-(4-chloropyrimidin-2-ylamino)benzonitrile (85 gm) as obtained in preparative example 1 and (E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile hydrochloride (69 gm) as obtained in preparative example 2 was added N- methylpyrrolidone (425 ml) under stirring. The mixture was then heated to 85 to 95C and stirred for 16 hours. The solution was then cooled to room temperature and then added water (1 105 ml). The reaction mass was stirred for 1 hour 30 minutes at room temperature and filtered. The solid obtained was then dried to obtain 135 gm of rilpivirine as a white solid.Chromatographic purity of rilpivirine: 98.6%;Content of Z-isomer: 1.2%.
135 g
In 1-methyl-pyrrolidin-2-one; at 85 - 95℃; for 16h;
Example 1 Preparation of rilpivirine To a mixture of 4-(4-chloropyrimidin-2-ylamino)benzonitrile (85 gm) as obtained in preparative example 1 and (E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile hydrochloride (69 gm) as obtained in preparative example 2 was added N-methylpyrrolidone (425 ml) under stirring. The mixture was then heated to 85 to 95 C. and stirred for 16 hours. The solution was then cooled to room temperature and then added water (1105 ml). The reaction mass was stirred for 1 hour 30 minutes at room temperature and filtered. The solid obtained was then dried to obtain 135 gm of rilpivirine as a white solid.
With N-Bromosuccinimide; In acetonitrile; at 20℃; for 4h;
N-bromosuccinimide (0.0393 mol) was added portion wise at room temperature to Intermediate I (0.0327 mol), the preparation of which has been described in WO-03/016306, in CH3CN (100 ml). The mixture was stirred at room temperature for 4 hours. The precipitate was filtered off, washed with CH3CN and dried yielding 10.08 g of the desired end product. The filtrate was evaporated and purified by column chromatography (eluent: CH2CI2 100; 35-70 mum). The pure fractions were collected, the solvent was evaporated and the residue was crystallized from CH3CN. Yielding : 2.4 g of Intermediate 2. The two fractions were collected. Total yield: 12.48 g of Intermediate 2 (86 %, melting point: > 250C).
86%
With N-Bromosuccinimide; In acetonitrile; at 20℃; for 4h;
N-bromosuccinimide (0.0393 mol) was added portion wise at room temperature to Intermediate I (0.0327 mol), the preparation of which is described in WO-03/016306, Ui CH3CN (IOO mI). The mixture was stirred at room temperature for 4 hours. The EPO <DP n="30"/>precipitate was filtered off, washed with CH3CN and dried yielding 10.08 g of the desired end product. The filtrate was evaporated and purified by column chromato? graphy (eluent: CH2CI2 100; 35-70 mum). The pure fractions were collected, the solvent was evaporated and the residue was crystallized from CH3CN. Yield : 2.4 g of Compound L The two fractions were collected. Yield: 12.48 g of Compound I (86 %, melting point: > 250C).
86%
With N-Bromosuccinimide; In CH3CN; at 20℃; for 4h;
Example Al: Preparation of intermediate 2 Intermediate 1 Intermediate 2N-bromosuccinimide (0.0393 mol) was added portion wise at room temperature to Intermediate L, the preparation of which has been described in WO-03/016306 (0.0327 mol) in CH3CN (100 ml). The mixture was stirred at room temperature for 4 hours. The precipitate was filtered off, washed with CH3CN and dried yielding 10.08 g of the desired end product. The filtrate was evaporated and purified by column chromatography (eluent: CH2Cl2 100; 35-70 mum). The pure fractions were collected, the solvent was evaporated and the residue was crystallized from CH3CN. Yielding : 2.4 g of Intermediate 2. The two fractions were collected. Yielding: 12.48 g of Intermediate 2 (86 %, melting point: > 250C).
86%
With N-Bromosuccinimide; In acetonitrile; at 20℃; for 4h;
Example 1 : Preparation of intermediate 2Intermediate 1 Intermediate 2N-bromosuccinimide (0.0393 mol) was added portion wise at room temperature to Intermediate 1 (0.0327 mol), the preparation of which is described in WO-03/016306, in CH3CN (100 ml). The mixture was stirred at room temperature for 4 hours. The precipitate was filtered off, washed with CH3CN and dried yielding 10.08 g of the <n="31"/>desired end product. The filtrate was evaporated and purified by column chromatography (eluent: CH2Cl2 100; 35-70 mum). The pure fractions were collected, the solvent was evaporated and the residue was crystallized from CH3CN. Yield : 2.4 g of Intermediate 2. The two fractions were collected. Total yield: 12.48 g of intermediate 2 (86 %, melting point: > 2500C).
With N-chloro-succinimide; In acetonitrile; at 20℃; for 4h;
N-chlorosuccinimide (0.000327 mol) was added portion wise at room temperature to Intermediate I (0.000273 mol) in CH3CN (5 ml). The mixture was stirred at room temperature for 4 hours. The precipitate was filtered, washed with CH3CN and dried. Yield: 0.065 g of intermediate 3 (59 %, melting point: > 250C).
59%
With N-chloro-succinimide; In CH3CN; at 20℃; for 4h;
Example A2: Preparation of intermediate 3 Intermediate 3 EPO <DP n="51"/>N-chlorosuccinimide (0.000327 mol) was added portion wise at room temperature to Intermediate 1 (0.000273 mol) in CH3CN (5 ml). The mixture was stirred at room temperature for 4 hours. The precipitate was filtered, washed with CH3CN and dried. Yielding: 0.065 g (59 %, melting point: > 2500C).
With Iodine monochloride; calcium carbonate; In ethanol; dichloromethane; water; at 20℃; for 24h;
A suspension Of CaCO3 (1.64g) in water (30ml) was added to a suspension of intermediate I (0.0273 mol) in EtOH (180ml). Iodine chloride (ICl) in CH2Cl2 (IN) (22.5ml) was added dropwise. The mixture was stirred at room temperature for 24 hours, then cooled to 0C and filtered. The filtrate was dried under vacuo, then taken up in EtOH (180ml), filtered, washed with EtOH and CH3CN and dried. Yield: 8.5g . Part of the filtrate was evaporated. The residue was crystallized from hot CH3CN. The precipitate was filtered off and dried. Yielding: 1.54g of intermediate 5 (total yield 78%).
78%
With Iodine monochloride; calcium carbonate; In ethanol; dichloromethane; water; at 20℃; for 24h;
A suspension Of CaCO3 (1.64g) in water (30ml) was added to a suspension of intermediate 1 (0.0273 mol) in EtOH (180ml). Iodine chloride (ICl) in CH2Cl2 (IN) (22.5ml) was added dropwise. The mixture was stirred at room temperature for 24 hours, then cooled to 0C and filtered. The filtrate was dried under vacuo, then taken up in EtOH (180ml), filtered, washed with EtOH and CH3CN and dried. Yield: 8.5g. Part of the filtrate was evaporated. The residue was crystallized from hot CH3CN. The precipitate was filtered off and dried. Yield: 1.54g (total yield 78%).
(E)-4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]pyrimidinyl]amino]benzonitrile tosylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In tetrahydrofuran; at 45℃;
<strong>[500287-72-9]Rilpivirine</strong> base form II (0.30 g, 0.82 mmol) was dissolved in THF (6 mL) at 45C. p-Toluenesulfonic acid monohydrate (171 mg, 0.90 mmol, 1.1 eq) was dissolved in THF (3 mL) at room temperature and the resulting solution was then added portion- wise to the solution containing <strong>[500287-72-9]Rilpivirine</strong> base at 45C with stirring. Precipitation was observed upon mixing. The reaction mixture was removed from the heat source and stirring was continued at room temperature for about 17 h. The precipitate was isolated by filtration to give <strong>[500287-72-9]Rilpivirine</strong> p-toluenesulfonate Form 1, as indicated by XRD.
1 g
In acetone; at 20℃; for 1h;
Example 11 Preparation of Tosylate Salt of <strong>[500287-72-9]Rilpivirine</strong> [0092] <strong>[500287-72-9]Rilpivirine</strong> (1 gm) was dissolved in acetone (50 ml) and then added a solution of p-toluene sulfonic acid (0.4 gm) in acetone (20 ml). The reaction mass was stirred for 1 hour at room temperature and filtered. The solid obtained was dried to give 1 gm of to sylate salt of <strong>[500287-72-9]rilpivirine</strong>.
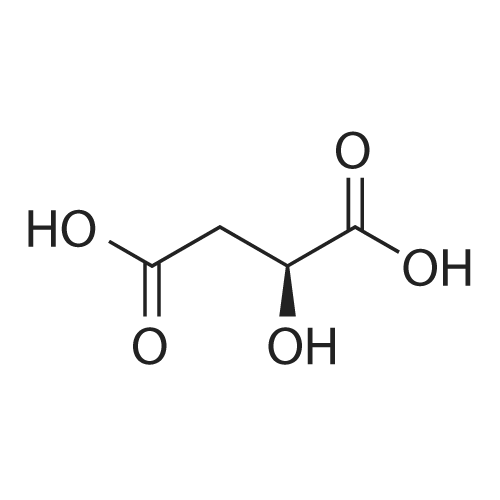
<strong>[500287-72-9]Rilpivirine</strong> base Form II (0.30 g, 0.82 mmol) was dissolved in THF (6 mL) at 45C. L-Malic acid (121 mg, 0.90 mmol, 1.1 eq) was dissolved in THF (3 mL) at room temperature and the resulting solution was then added portion- wise to the solution containing <strong>[500287-72-9]Rilpivirine</strong> base at 45C with stirring. The reaction mixture was removed from the heat source and stirring was continued at room temperature for about 17 h. A precipitate formed and was isolated by filtration to give <strong>[500287-72-9]Rilpivirine</strong> L-malate Form 1. Drying of this material in a vacuum oven at 50C for 18 h afforded the same polymorphic form as was identified prior to drying.
<strong>[500287-72-9]Rilpivirine</strong> base form II (0.30 g, 0.82 mmol) was dissolved in THF (6 mL) at 45C. L-tartaric acid (135 mg, 0.90 mmol, 1.1 eq) was dissolved in THF (3 mL) at roomtemperature and the resulting solution was then added portion- wise to the solution containing <strong>[500287-72-9]Rilpivirine</strong> base at 45C with stirring. The reaction mixture was removed from the heat source and stirring was continued at room temperature for about 17 h. The mixture was then cooled to -18C and maintained at this temperature overnight, and then returned to ambient temperature. To the mixture was then added diethyl ether (6 mL), leading to the formation of a precipitate. The precipitate was isolated by vacuum filtration and the filter cake was washed with diethyl ether to give amorphous <strong>[500287-72-9]Rilpivirine</strong> L-tartrate as a white solid, both before and after drying in a vacuum oven at 60C overnight. The samples were found to be amorphous.
In tetrahydrofuran; at 45℃;Product distribution / selectivity;
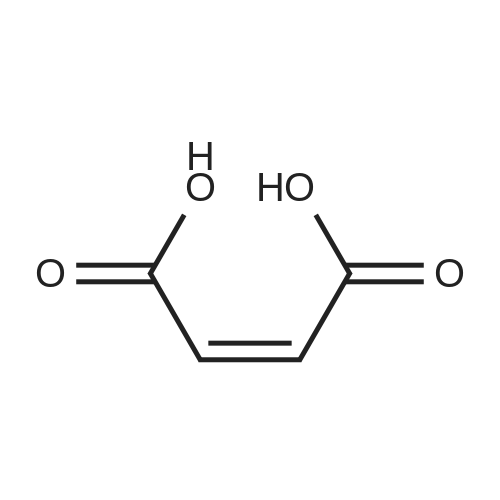
<strong>[500287-72-9]Rilpivirine</strong> base Form II (0.30 g, 0.82 mmol) was dissolved in THF (6 mL) at 45C. Maleic acid (105 mg, 0.90 mmol, 1.1 eq) was dissolved in THF (3 mL) at room temperature and the resulting solution was then added portion- wise to the solution containing <strong>[500287-72-9]Rilpivirine</strong> base at 45C with stirring. The reaction mixture was removed from the heat source and precipitation was observed after stirring for about 20 min. The stirring was continued at room temperature for about 17 h. The precipitate was isolated by filtration to give <strong>[500287-72-9]Rilpivirine</strong> maleate form 1. Drying of this material in a vacuum oven at 50C for 17 h afforded the same polymorphic form as was identified prior to drying, as confirmed by XRD analysis.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping