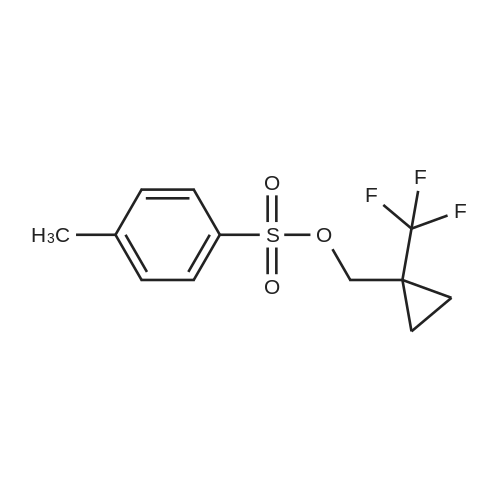
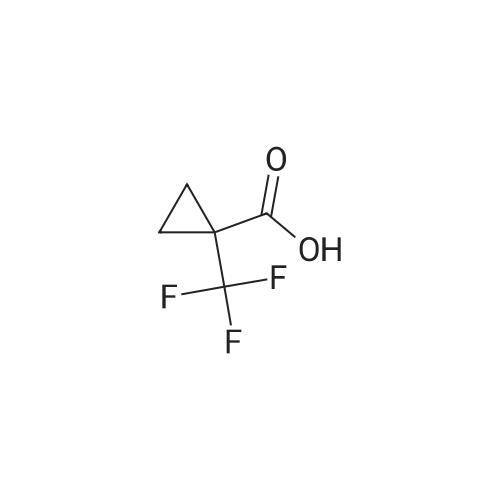
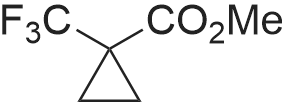
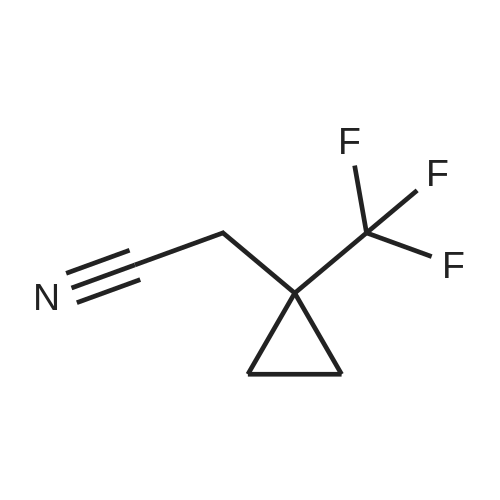
| 86% |
With lithium aluminium hydride; In diethyl ether; at 0 - 20℃;Inert atmosphere; |
Treat a 0C solution of l-(trifluoromethyl)cyclopropanecarboxylic acid (9 g, 58.4 mmol) in ether (140 mL), under N2, portion-wise with LAH (2.9 g, 76 mmol), allow to warm to RT and stir overnight. Re-cool to 0C, slowly add HC1, warm to RT and separate the layers. Extract the aqueous layer with ether (2x), wash the combined organics with brine, dry over Na2S04 and concentrate under reduced pressure (water bath temp <30C) to afford the title compound (7 g, 86%). 1HNMR (400 MHz, DMSO-d6): δ 4.94 (t, J = 6.0 Hz, 1 H), 3.54 (d, J = 6.0 Hz, 2 H), 0.87-0.84 (m, 2 H), 0.81-0.79 (m, 2 H). |
| 77% |
|
Step 1: 50 mL of borane dimethylsulfide 2M in THF were added dropwise at RT via an addition funnel to a solution of 1-trifluormethyl-cyclopropanecarboxylic acid (10 g, 64.90 mmol) in 100 mL of THF. The resulting clear solution was stirred at 40 C. for 24 hours before being cooled to 0 C. and quenched by slow addition of aqueous saturated NH4Cl. The biphasic slurry was filtered through celite. The organic layer was separated, and the aqueous layer was back extracted twice with AcOEt. The combined organic layers were dried (Na2SO4), filtered, and evaporated to give 7 g of (1-trifluoromethyl-cyclopropyl)-methanol (77% yield). Used as in the next step. |
| 77% |
|
A mixture of 1-(trifluoromethyl)cyclopropane-1-carboxylic acid (0.50 g, 3.25 mmol) BH3 (1 N in THF, 5 mL) was refluxed for 1 hr and cooled to room temperature. 1 mL 12 N aqueous HCl was added. The reaction mixture was stirred for another 10 min. The solvent was removed under reduced pressure. The residue was partitioned between Et2O (20 mL) and water (20 mL). The organic layer was separated. The aqueous layer was extracted with Et2O (2 x 20 mL). The combined organic layer was washed with brine (2 x 50 mL), dried over anhydrous Na2SO4and evaporated to give the product as a colorless oil (0.35 g, yield: 77%). |
| 70% |
With lithium aluminium hydride; In diethyl ether; at 0 - 20℃; |
l-(Trifluoromethyl)cyclopropane-l-carboxylic acid (858 mg, 5.57 mmol, 1.00 eq.) was dissolved in diethyl ether (15 mL). The reaction mixture was cooled to 0 C. Lithium aluminum hydride (274 mg, 7.24 mmol, 1.30 eq.) was added portionwise. The reaction mixture was stirred overnight and allowed to reach room temperature. The reaction mixture was cooled to 0 C. HC1 (aq., 1 N, 25 mL) was added dropwise. The aqueous phase was extracted with diethyl ether (2 χ 25 mL). The combined organic phases were washed with brine (25 mL), dried over sodium sulfate, filtered and evaporated in vacuo (Tbath < 30 C) to give (l-(trifluoromethyl)cyclopropyl)methanol (547 mg, 3.90 mmol, 70% yield) as a colorless oil. 1H NMR (CDC13): d 3.73 (s, 2H), 1.58 (br, 1H), 1.07-1.01 (rn 2H), 0.82-0.75 (m, 2H). |
| 45% |
With lithium aluminium hydride; In diethyl ether; at 0℃; for 2h;Inert atmosphere; |
Lithium aluminum hydride (183mg, 4.8mmol) was dissolved in anhydrous diethyl ether (10 mL), and replaced with nitrogen. Was cooled to 0 deg. C, was slowly added ether solution of 1-trifluoromethyl-1-cyclobutane carboxylic acid. At this temperature the reaction 2 hours, 0.5ml of saturated sodium sulfate solution was added, inorganics filtered, dried, and concentrated to give [1-(trifluoromethyl)cyclopropyl]methanol (pale yellow liquid, 250mg), 45% yield. |
| 45.3% |
With lithium aluminium hydride; In tetrahydrofuran; at 0 - 40℃; |
Step 1 1-(Trifluoromethyl)cyclopropane-1-carboxylic acid (XXV) (3.7334 g, 24.23 mmol) was dissolved in THF (162 mL) and cooled to 0 C. LAH (1.1614 g, 29.07 mmol) was then added and the reaction heated to 40 C. overnight. The reaction was cooled to 0 C. Water (2 mL) was added to quench the reaction followed by 2 N NaOH (0.3 mL). The reaction was stirred forming a precipitate which was filtered off and washed with ether. The aqueous phase was removed and the organic phase was washed with brine, dried, and carefully concentrated to give (1-(trifluoromethyl)cyclopropyl)methanol (XXVI) (1.5376 g, 10.98 mmol, 45.3% yield) as a clear, volatile liquid. |
| 45.3% |
With lithium aluminium hydride; In tetrahydrofuran; at 0 - 40℃; |
1-(Trifluoromethyl)cyclopropane-1-carboxylic acid (LVIII) (3.7334 g, 24.23 mmol) was dissolved in THF (162 mL) and cooled to 0 C. LAH (1.1614 g, 29.07 mmol) was then added and the reaction heated to 40 C. overnight. The reaction was cooled to 0 C. Water (2 mL) was added to quench the reaction followed by 2 N NaOH (0.3 mL). The reaction was stirred forming a precipitate which was filtered off and washed with ether. The aqueous phase was removed, and the organic phase was washed with brine, dried, and carefully concentrated to give (1-(trifluoromethyl)cyclopropyl)methanol (LIX) (1.5376 g, 10.98 mmol, 45.3% yield) as a clear, volatile liquid. |
| 28.6% |
With lithium aluminium hydride; In tetrahydrofuran; at 20℃; |
A 2M solution of lithium aluminum hydride in THF (2.11 mL, 4.22 mmol) was added dropwise to a solution of 1-(trifluoromethyl)-1-cyclopropanecarboxylic acid (500.0 mg, 3.24 mmol) in THF (8 mL) cooled to 0C The reaction mixture was stirred at room temperature overnight then was quenched with sodium sulfate decahydrate. The resulting suspension was filtered and the filtrate was evaporated in vacuo to give [1- (trifluoromethyl)cyclopropyl]methanol (130 mg, 0.928 mmol, 28.6% yield) as a colourless oil.1H NMR (400 MHz, DMSO-d6) δ 0.75 - 0.92 (m, 4H), 3.55 (d, J = 5.91 Hz, 2H), 4.93 (t, J = 6.00 Hz, 1H). |
| 28.6% |
With lithium aluminium hydride; In tetrahydrofuran; at 20℃; |
A 2M solution of lithium aluminum hydride in THF (2.11 mL, 4.22 mmol) was added dropwise to a solution of 1-(trifluoromethyl)-1-cyclopropanecarboxylic acid (500.0 mg, 3.24 mmol) in THF (8 mL) cooled to 0C The reaction mixture was stirred at room temperature overnight then was quenched with sodium sulfate decahydrate. The resulting suspension was filtered and the filtrate was evaporated in vacuo to give [1- (trifluoromethyl)cyclopropyl]methanol (130 mg, 0.928 mmol, 28.6% yield) as a colourless oil.1H NMR (400 MHz, DMSO-d6) δ 0.75 - 0.92 (m, 4H), 3.55 (d, J = 5.91 Hz, 2H), 4.93 (t, J = 6.00 Hz, 1H). |
|
With lithium aluminium hydride; In diethyl ether; for 1h;Inert atmosphere; |
Step 1: Synthesis of (l-(trtfluoromethyl)cyclopropyl)methanol:To a stirred solution of Lithium aluminium hydride (2.4 g) in dry ether (70 ml) l-(trifluoromethyl)cyclopropanecarboxylic acid (5 g) was added at 0 C under N2 atmosphere and the reaction mixture was stirred for about 1 hour. After completion of the reaction (monitored by - NMR), the reaction mixture was quenched by cautious addition of water (1 ml), 15 % aq. NaOH solution (1 ml) and again water (3 ml) and stirred for 30 minutes. The reaction mixture was filtered and cake was washed with ether twice. Due to volatile nature of the compound, only half of the ether volume was reduced on hot water both using without any vacuum and next reaction was proceeded as such. 1H NMR (CDC13, 300 MHz): δ 3.73 (s, 2H), 1.83 (br s, 1H), 1.05- 1.01 (m, 2H), 0.80- 0.76 (m, 2H); ES Mass: (M- H) 138.91 |
|
|
To a solution of compound 1-trifluoromethyl-cyclopropanecarboxylic acid (2 g, 13 mmol) in dry THF (80 mL) was added LAH (592 mg, 16 mmol) in portions at 0 C. and the resulting mixture was heated at 40 C. overnight. H2O (592 mg, 16 mmol) was added to quench the reaction at 0 C. and followed by 2N NaOH (0.6 mL). After filtration, the filtrate was distilled to remove the most solvent to give crude (1-trifluoromethyl-cyclopropyl)-methanol (1.2 g crude), which was used in the next step without further purification. |
|
With lithium aluminium hydride; In tetrahydrofuran; at 30℃; for 14h;Cooling with ice; |
Lithium aluminum hydride (approximately 78.45 g, 2.067 mol) (pellets) were added to the flask, THF (2.450 L) was added to the addition funnel, and the system was cycled 3 times with vacuum/ nitrogen. The solvent was quickly added to the LAH pellets, stirred at room temperature for 0.5 h (pellets start to fall apart to give a grey suspension), and cooled in an ice bath. A solution of 1-(trifluoromethyl)cyclopropanecarboxylic acid (245 g, 1.590 mol) in THF (735.0 mL) was slowly added via an addition funnel over 0.5-1 h, keeping the internal temperature below 30 C. The grey suspension was stirred in the melting ice bath for 14 hours. The grey suspension was quenched under ice cooling by slow addition of water (approximately 75.92 g, 75.92 mL, 4.214 mol), followed by NaOH (approximately 76.32 mL of 6 M, 457.9 mmol) and water(approximately 75.92 g, 75.92 mL, 4.214 mol). The grey suspension was stirred at ~50 C till the solid became colorless (~0.5 h), treated with magnesium sulfate (20 g), filtered over Celite, and the aluminium salts were washed with three portions of hot THF. The filtrate was dried again over magnesium sulfate, filtered, and concentrated by evaporation at 55 C and 450 mbar to give [1-(trifluoromethyl)cyclopropyl]methanol as a 62 wt% solution (NMR) in THF (327 g, 91%). 1H NMR (400 MHz, DMSO-d6) δ 4.94 (t, J = 6.0 Hz, 1H), 3.56 (d, J = 6.0 Hz, 2H), 0.91 - 0.74 (m, 4H) |
|
With lithium aluminium hydride; In tetrahydrofuran; diethyl ether; at -15℃; for 0.5h; |
1 M Lithium aluminium hydride in tetrahydrofuran (4.55ml, 4.55mmol) was slowly added to a stirred solution of 1-(trifluoromethyl)cyclopropanecarboxylic acid (1g, 6.5mmol) in diethyl ether (6ml) at -15C. The mixture was stirred for 30 minutes, warmed to room temperature, quenched with water and extracted with diethyl ether (x3). The combined organic phases were dried (MgS04) and concentrated to ~5ml. |
|
With lithium aluminium hydride; In tetrahydrofuran; at 26℃; for 18h; |
To a round bottom flask were added 1- (trifluoromethyl) cyclopropanecarboxylic acid (5000 mg, 32.4 mmol) , THF (100 mL) and LiAlH4(1847 mg, 48.7 mmol) in portions at 0 . The reaction mixture was stirred for 18 h at 26 . The mixture was cooled to RT, quenched with 15NaOH solution (1.85 mL) and water (1.85 mL) . Anhydrous sodium sulfate was added and the mixture was stirred for 30 mins. The mixture was filtered and the filtrated was concentrated in vacuum to give the title compound.1H NMR (CDCl3, 400 MHz) : δ 4.92 (t, J 6.06 Hz, 1H) , 3.52 (d, J 5.87 Hz, 2H) , 0.80-0.86 (m, 2H) , 0.73-0.80 (m, 2H) . |
| 5.11 g |
With dimethylsulfide borane complex; In tetrahydrofuran; at 40℃; for 18h;Inert atmosphere; |
To a stirred solution of 1-(trifluoromethyl)cyclopropane-1-carboxylic acid (6.0 g, 38.96 mmol) in anhydrous tetrahydrofuran (35 mL) was added borane-methyl sulfide complex (29.2 mL, 2.0 M solution in THF, 58.4 mmol) at room temperature under argon atmosphere. The resulting reaction mixture was stirred at 40 C for 18 h. The reaction was quenched by adding saturated aqueous ammonium chloride solution (120 mL). The resulting solid was filtered off. The filtrate was extracted with diethyl ether (50 mL × 3). The combined organic solution was washed with saturated aqueous sodium bicarbonate solution (100 mL) and brine (100 mL). Then, the organic solution was dried over anhydrous sodium sulfate, filtered and concentrated under vacuum to give (1-(trifluoromethyl)cyclopropyl)methanol as a light yellow oil (5.11 g). LCMS: MS (ESI) m/z was not observed. 1H NMR (400 MHz, chloroform-d) δ 3.73 (s, 2H), 1.05-1.02 (m, 2H), 0.78 (m, 2H) ppm. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +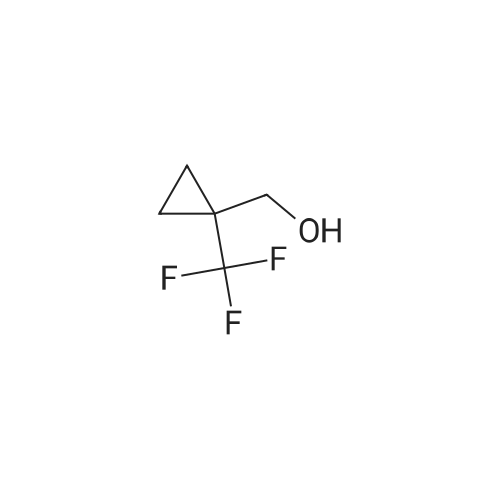

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping