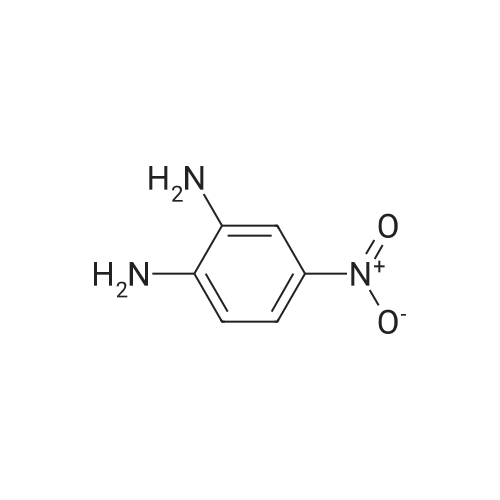
| 94% |
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 0 - 20℃; |
To a solution of compound M-1 (2 g, 7.2 mmol) and M-2 (1.5 g, 6.55 mmol) in MeCN (50 mL) was added DIPEA (1.3 mL) at 0 °C. At the end of the addition, the mixture was stirred at rt and the reaction was monitored by TLC. After the reaction was completed, the mixture was quenched with ice water (10 mL) and extracted with EtOAc (100 mL x 3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc (v/v) = 4/1) to give compound M-3 (2.55 g, 94percent). The compound was characterized by the following spectroscopic data: 1H NMR (400 MHz, CDCl3): delta 7.78 (m, 2H), 7.65 (m, 2H), 5.54-5.15 (m, 2H), 4.39 (m, 1H), 3.76 (m, 1H), 3.03 (m, 1H), 2.53 (m, 1H), 2.27 (m, 1H), 1.84 (m, 1H), 1.43 (m, 9H), 1.24 (m, 1H), 1.09 (m, 3H). [0524] A mixture of compound M-3 (2.55 g, 6.2 mmol) and ammonium acetate (4.6 g, 59.7 mmol) in xylene (100 mL) in a sealed tube was stirred at 130 °C for 5 hours, cooled to rt and washed with water. The organic layers were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc (v/v) = 4/1) to give compound M-4 (1.63 g, 64.7percent). The compound was characterized by the following spectroscopic data: 1H NMR (400 MHz, CDCl3): delta 7.78 (m, 2H), 7.65 (m, 2H), 4.39 (m, 1H), 3.76 (m, 1H), 3.03 (m, 1H), 2.53 (m, 1H), 2.27 (m, 1H), 1.84 (m, 1H), 1.43 (m, 9H), 1.24 (m, 1H), 1.09 (m, 3H). [0525] To a mixture of compound M-4 (1.63 g, 4 mmol), bis(pinacolato)diboron (1.12 g, 4.4 mmol), Pd(dppf)Cl2·CH2Cl2 (71 mg, 0.09 mmol) and KOAc (0.98 g, 10 mmol) was added DME (20 mL) under N2 via syringe. The resulting mixture was stirred at 90 °C for 5 hours in an oil bath and concentrated in vacuo. To the residue was added a small amount of water, and the mixture was extracted with EtOAc (30 mL x 3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc (v/v) = 2/1) to give the title compound 28-1 as a pale yellow solid (1.77 g, 97.6percent). The compound was characterized by the following spectroscopic data: 1H NMR (400 MHz, CDCl3): delta 7.78 (m, 2H), 7.65 (m, 2H), 4.39 (m, 1H), 3.76 (m, 1H), 3.03 (m, 1H), 2.53 (m, 1H), 2.27 (m, 1H), 1.84 (m, 1H), 1.43 (m, 9H), 1.24 (m, 1H), 1.09 (m, 3H). |
|
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 20℃; for 5h; |
To a solution of M12.2c (1.38 g, 6.02 mmol) and 2-bromo-l-(4- bromophenyl)ethanone (1.673 g, 6.02 mmol) in acetonitrile (35 mL) was added DIEA (1.051 mL, 6.02 mmol). It was stirred at room temperature for 5 hrs. The solvent was evaporated in vacuo and water (50 mL) and EtOAc (70 mL) were added, organic layer was separated and washed by sat. aHC03 (30 mL), dried with Na2S04, evaporated in vacuo to give crude M12.2d as a red oil (2.71 g), which was used in the next step without further purification. XH NMR (DMSO-d6, delta = 2.5 ppm, 400 MHz): 7.91 (m, 2H), 7.78 (d, J = 8.5 Hz, 2H), 5.60-4.90 (m, 2H), 4.29 (app t, 1H), 3.61 (m, 1H), 2.85 (m, 1H), 2.35-1.80 (m, 2H), 1.65 (m, 1H), 1.40-1.32 (two s, 9H), 1.02 (d, J = 6.5 Hz, 3H). LC/MS: Anal. Calcd. for [M+Na Ci9H24 81BrNNa05: 450.07; found: 450.11. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping