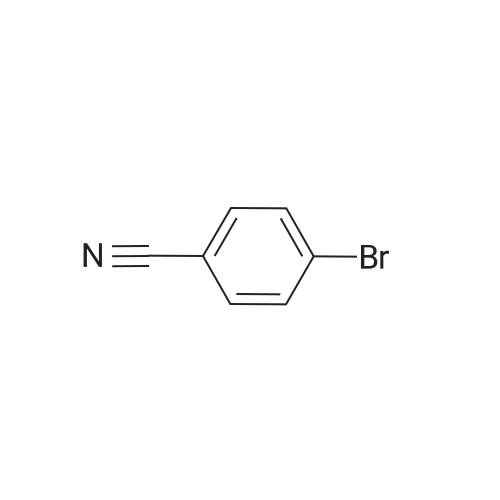
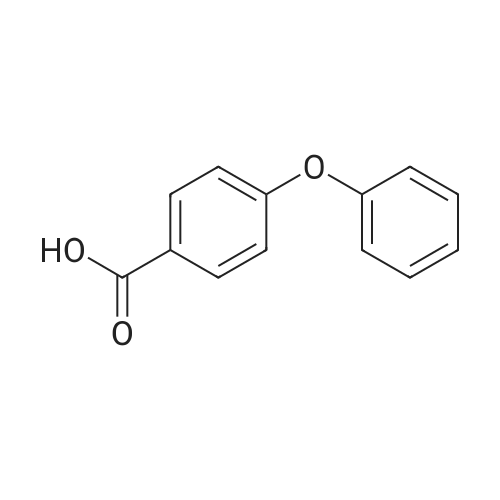
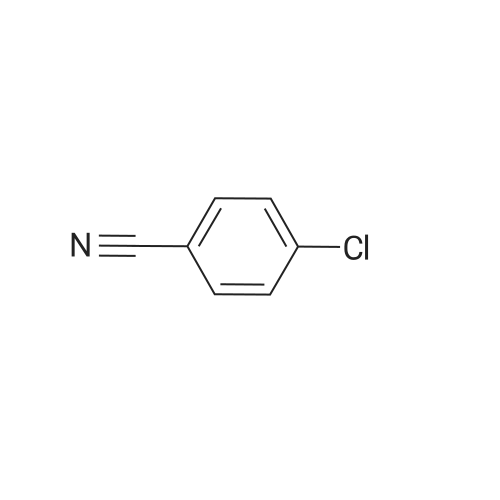
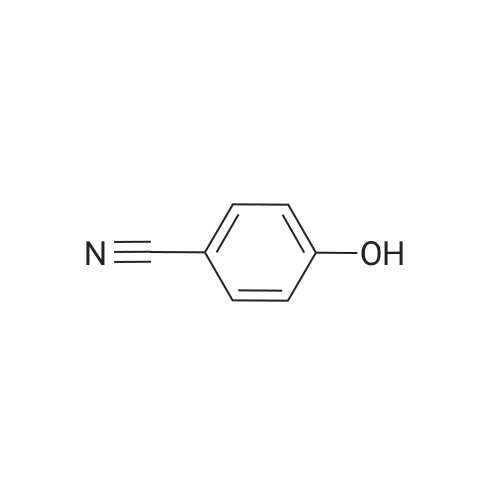
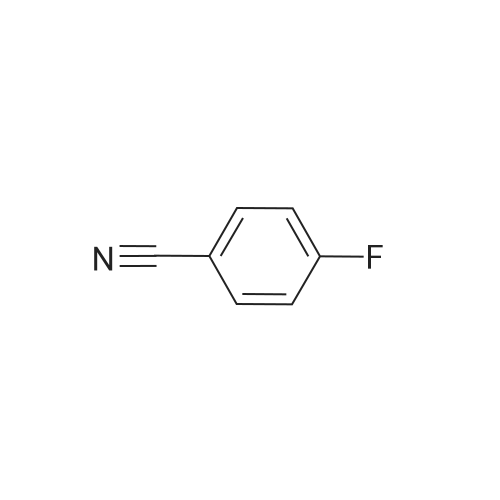
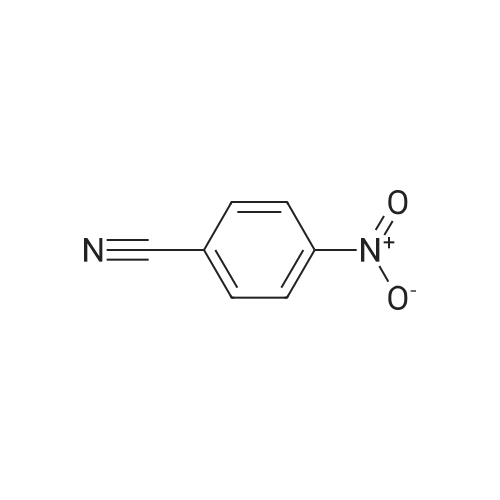
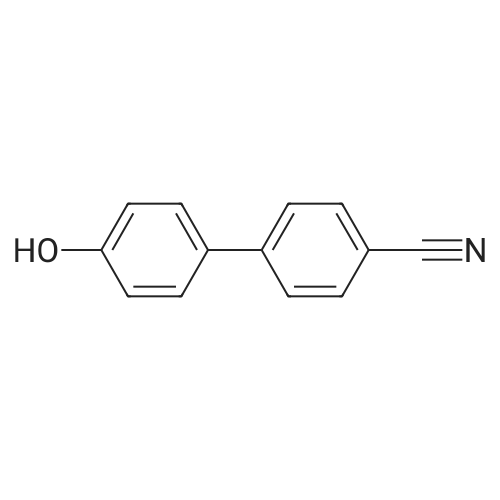
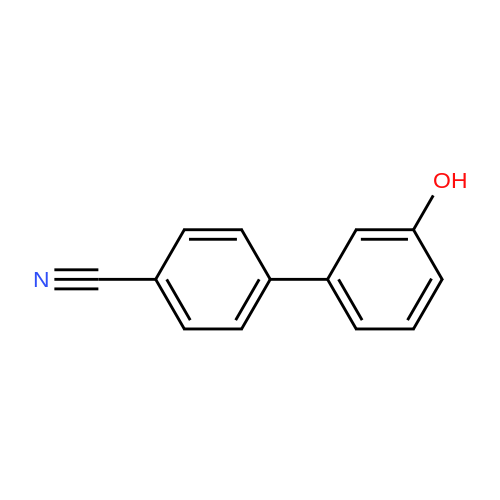
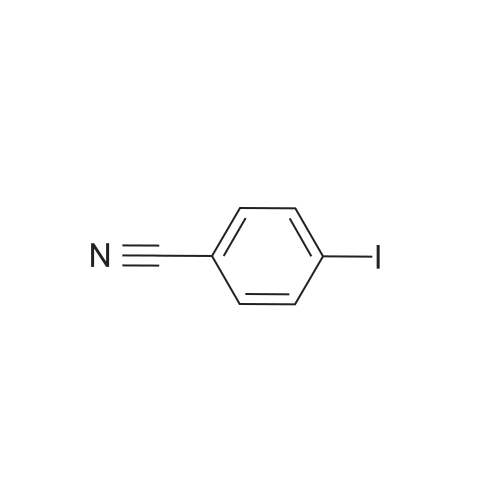
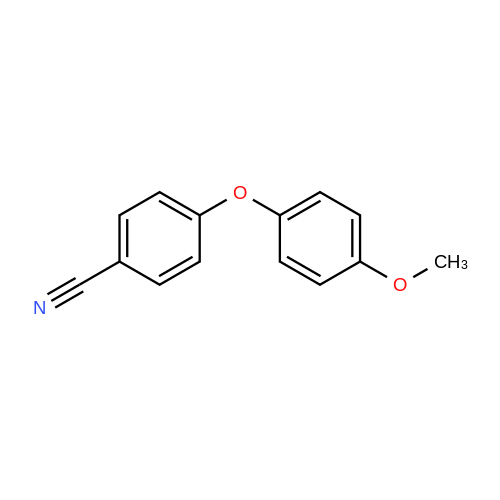
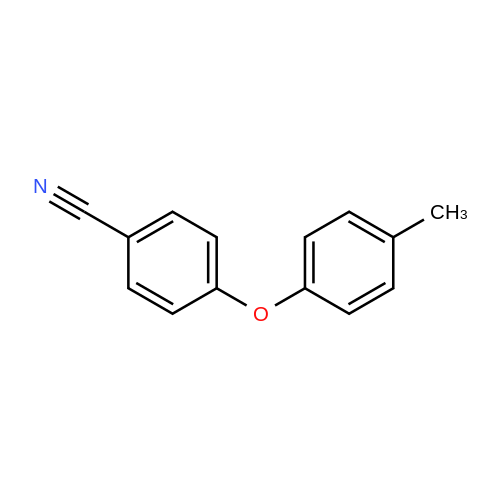
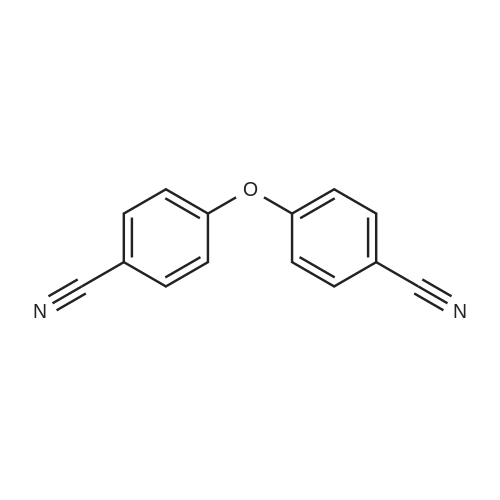
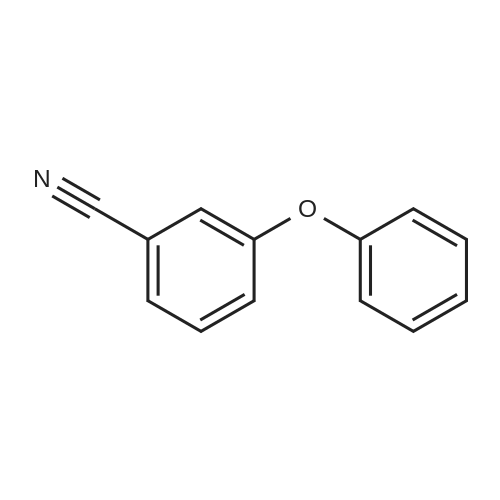
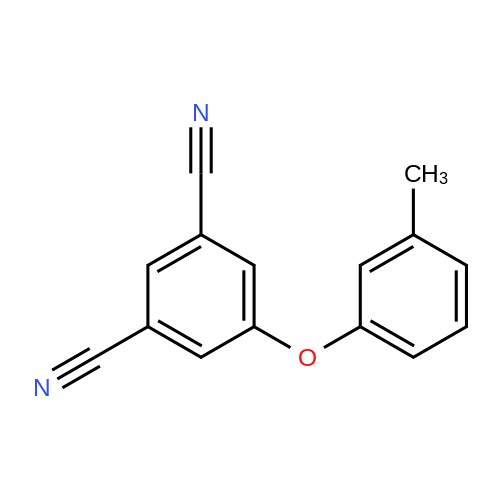
| 87% |
With copper(II) oxide; potassium hydroxide; In N,N-dimethyl acetamide; at 27℃; for 24h;Inert atmosphere; Sealed tube; |
General procedure: A magnetic stirring bar, nanocrystalline CuO (10 mg, 3 mol %), KOH (112 mg, 2 mmol) and phenol/substituted phenol/ thiophenol (1.2 mmol) were added into an oven-dried flask (25 mL). The flask was sealed with a septum, followed by three cycles of evacuation and filling with dry nitrogen. Then aryl halide (1 mmol) and N,N-dimethyl acetamide (DMAc) (4 mL) were injected through a syringe. The flask was sealed and stirred under nitrogen until the completion of the reaction (as monitored by TLC or GC). The catalyst was recovered from the reaction mixture and washed several times with ethyl acetate. The catalyst-free reaction mixture was quenched with brine solution and the product was extracted with ethyl acetate. The combined organic extracts were dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated and the residue was purified by column chromatography on silica gel (hexane/ethyl acetate, 80/20) to afford the product with high purity. |
| 86% |
With tetrabutylammonium bromide; Cs2CO3; In dimethyl sulfoxide; at 120℃; for 12h; |
General procedure: Polymer supported Cu(II) catalyst (0.05 g, 0.0098 mmol) in DMSO (5 mL) was taken in a 100 ml R.B flask and stirred at room temperature for 10 min. Then aryl halide (1 mmol), phenol(1 mmol), tetrabutylammonium bromide (tBu4NBr) (0.1 mmol),Cs2CO3 (1 mmol) and DMSO (5 mL) were added to it. The final reaction mixture was refluxed at 120 C under an open air condition.The reaction mixtures were collected at different time intervals and identified by GCMS and quantified by GC. After the completion of the reaction, the catalyst was filtered off and washed with water followed by acetone and dried in oven. The filtrate was extracted with ethyl acetate (3 x 20 ml) and the combined organic layers were dried with anhydrous Na2SO4 by vacuum. The filtrate was concentrated by vacuum and the resulting residue was purified by column chromatography on silica gel to provide the desired product. |
| 86% |
With thio-xanthene-9-one; (2,2'-bipyridine)nickel(II) dibromide; N-tert-butylisopropylamine; In acetonitrile; for 72h;Inert atmosphere; Irradiation; Green chemistry; |
p-bromobenzonitrile (0.2 mmol), phenol (0.6 mmol), TXO (20 mol %), Ni(bpy)Br2 (10 mol %), t-BuNH(i-Pr) (0.4 mmol) and MeCN ( 3 mL) was added to a dry reaction tube with a magnetic stirrer, then the reaction tube was replaced with N2 3 times, and the reaction was stirred for 72 hours under 45 W CFL irradiation. After the reaction, 5 mL of water was added, then extracted with 3 × 5 mL of ethyl acetate, the organic phases were combined, and the organic phases were dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated by rotary evaporation and separated by silica gel column chromatography. The target product was obtained (yield 86%). |
| 76% |
With [NiII(N-((6-fluoropyridin-2-yl)methyl)(pyridin-2-yl)-N-(pyridin-2-ylmethyl)methanamine)(acetate)(H2O)][BPh4]; potassium carbonate; In dimethyl sulfoxide; at 120℃; for 10h;Catalytic behavior; |
General procedure: In a round bottom flask, mixture of phenol (1.2 mmol), aryl halides(1 mmol) and K2CO3 (2 mmol) have been taken in 5 mL DMSO. Thecomplexes (1-3) (4 mol%) were then added into the reaction mixture.Thereafter, the reaction mixture was heated at 120 C for the given time.The progress of reaction was monitored through TLC made of silica andafter the maximum conversion reached, reactions were quenched bywater after cooling and extracted using ethyl acetate. Then, it was driedover anhydrous sodium sulphate and subjected to GC analysis usingShimadzu GC-2014. The products were detected and quantified by GC(FID) with the following temperature program: injector temperature240 C; initial temperature 50 C, isothermal for 3 min then heating rate10 C min 1 to 200 C and thereafter another isothermal of 3 min; FIDtemperature 250 C. |
| 74% |
With potassium-t-butoxide; In dimethyl sulfoxide; at 120℃; for 2h;Inert atmosphere; |
General procedure: A 25-mL Schlenk tube was flame-dried under vacuum and filled with argon after cooling to room temperature. To this tube were added phenol (1.0 mmol), t-BuOK (2.0 mmol). The tube was then evacuated and backfilled with argon (3 cycles). A dry DMSO solution (1.0 mL) of aryl bromides(2.0 mmol) was loaded into a plastic syringe. After the tube was purged with argon, this solutionwas injected into bottom of the tube using a long needle syringe. The mixture was stirred under Ar atmosphere in sealed Schlenk tubes at the corresponding temperature. When the reaction was cooled down to room temperature, the mixture was filtered through a short plug of silica gel and washed with 100 mL dichloromethane and water. The combined organic phase was concentrated under vacuum. The product was purified through flash column chromatography on 200-300 mesh silica gel with petroleum ether/ethyl acetate as eluent with a suitable ratio according to the TLC experiments. The identity and purity of the product were ascertained by GC-MS, HRMS, 1H and 13C NMR spectroscopy. |
| 70% |
With tetrabutylammonium bromide; Cs2CO3; In dimethyl sulfoxide; at 120℃; for 12h; |
General procedure: Cu- catalyst (0.05 g) in DMSO (5 mL) was taken in a 100 mLround bottom flask and stirred at room temperature for 10 min.Then aryl halide (1 mmol), phenol (1 mmol), tetrabutylammoniumbromide (tBu4NBr) (0.1 mmol), Cs2CO3(1 mmol) and DMSO (5 mL)were added to it. The final reaction mixture was heated at 120Cunder an open air condition. The reaction mixtures were collectedat different time intervals and identified by GC-MS and quantifiedby GC. After the completion of the reaction, the catalyst was fil-tered off and washed with water followed by acetone and dried inoven. The filtrate was extracted with ethyl acetate (3 × 20 mL) andthe combined organic layers were dried with anhydrous Na2SO4byvacuum. The filtrate was concentrated by vacuum and the result-ing residue was purified by column chromatography on silica gelto provide the desired product. |
| 70% |
With copper (I) iodide; 2-(2-benzoylhydrazine-1-carbonyl)-1-benzylpyrrolidine 1-oxide; Cs2CO3; In acetonitrile; at 80℃; for 12h;Inert atmosphere; Sealed tube; |
General procedure: CuI (19.2 mg, 0.1 mmol, 10 mol%), Cs2CO3 (650 mg, 2.0 mmol) and L2 (34 mg, 0.1 mmol, 10 mol%) were added to a re-sealable 25 mL test tubes with Teflon septa. The tube was evacuated and backfilled three times with nitrogen. Add the corresponding solvent (EtOH or CH3CN, 1 mL) via syringe under countercurrent nitrogenflow, continue adding halides (1.5 mmol) and nucleophile (1.0 mmol), seal the test tube. The reaction mixture was heated at 80 C for 12 h, and then allowed to cool to room temperature. After the reaction mixture was diluted with CH2Cl2, the precipitate was removed by filtration and washed with water. After extraction, the organic phase was dried over anhydrous sodium sulfate and concentrated by rotary evaporation. The residue was purified by column chromatography. |
| 65% |
With Cs2CO3; copper(II) bromide; 1,1'-azobis(1-cyanocyclohexanenitrile); In N,N-dimethyl-formamide; at 100℃; for 1.5h;Microwave irradiation; Green chemistry; |
General procedure: a mixture of aryl halide (1 mmol), unsubstituted or substituted phenol (1 mmol), Cs2CO3 (1 mmol), ACHN (0.2 mmol %), CuBr (0.2 mmol %) in DMF (10 mL) were added to the microwave tube. The reaction was found to be stable to air and DMF was used directly without purification. The mixture was reacted in a microwave oven at 100 C (extern temperature) for 30-60 min and the progress of the reaction was monitored byTLC. After completion of the reaction, it was allowed to reach room temperature; 10 mL of water was added and then the crude was extracted with ethyl ether (3 15 mL). Organic layer was washed with water (2 15 mL), dried over Na2SO4 and filtered. After removal of the solvent, the diaryl ether was isolated by silica gel column chromatography. |
| 59% |
With tris(dibenzylideneacetone)dipalladium(0) chloroform complex; di-tert-butyl(2′,4′,6′- triisopropyl-3,6-dimethoxy-[1,1′-biphenyl]-2-yl)phosphine; Cs2CO3; In neat (no solvent); at 110℃; for 18h;Sealed tube; Inert atmosphere; |
General procedure: An oven-dried screw-cap tube was cooled to roomtemperature under argon pressure and was charged with aryl halide(1.0 mmol), phenol (1.2 mmol), the ligand (2 mol%) and Pd2(dba)3·CHCl3(10 mg, 1 mol%). The mixture was well homogenized followed by additionof Cs2CO3 (650 mg, 2.0 mmol). The tube was sealed and placed in a preheatedoil bath at 110 C and stirred for 18 h. The mixture was thenallowed to cool to room temperature, and the resulting dark heterogeneousmixture was treated with water (10 ml) and ethyl acetate (10 ml). Theorganic phase was collected, filtered through a small pad of Celite andconcentrated under reduced pressure. The crude product was purifiedby flash chromatography on silica gel. |
|
With C25H22Cl2NPPd; potassium carbonate; In dimethyl sulfoxide;Reflux;Catalytic behavior; |
General procedure: A mixture of aryl halide (1.0 mmol), phenol (1.2 mmol), potas- sium carbonate (2.0 mmol), catalyst (0.1 mol%) and 4.0 mL of dimethylsulfoxide (DMSO) was taken in an oven dried round bot- tom flask of 100 mL capacity and refluxed at 110 C. The progress of the reaction was monitored through TLC. When the stage of the maximum conversion reached, the mixture was cooled to room temperature. The mixture was extracted using ethyl acetate and washed three times with water. The organic layer was made free from water using anhydrous sodium sulphate. Finally, the solvent was evaporated to obtain the product. The conversion has been es- timated using 1 H NMR study. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping