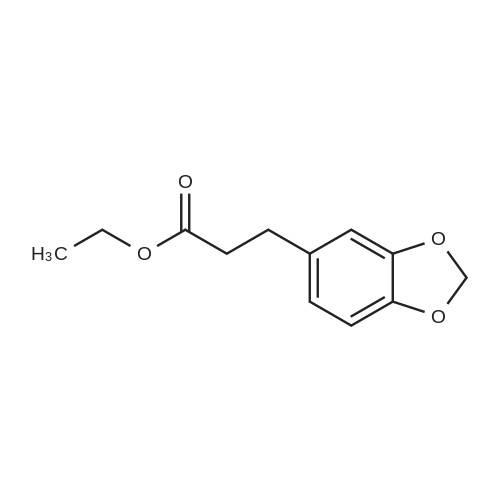
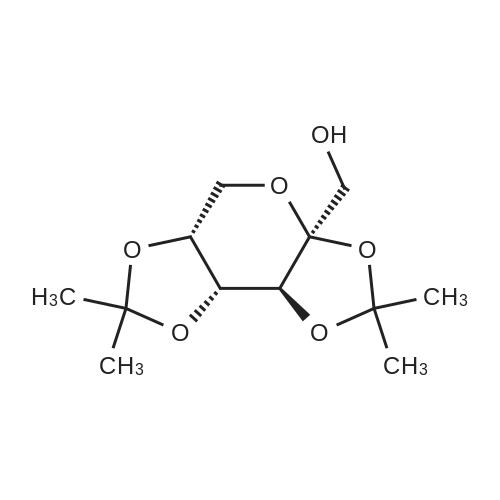
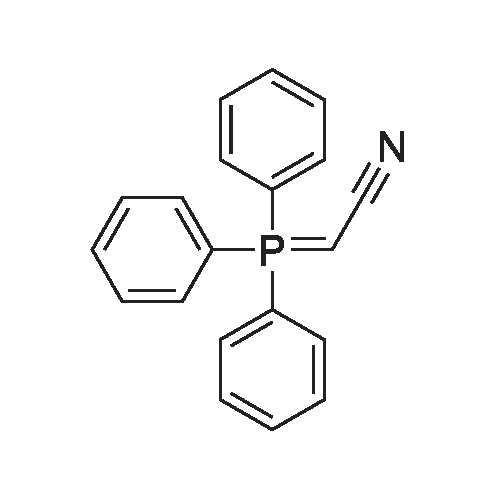
|
With thionyl chloride; at 79℃; for 4h; |
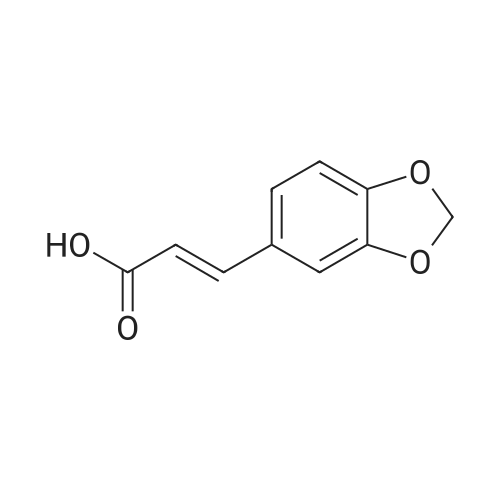
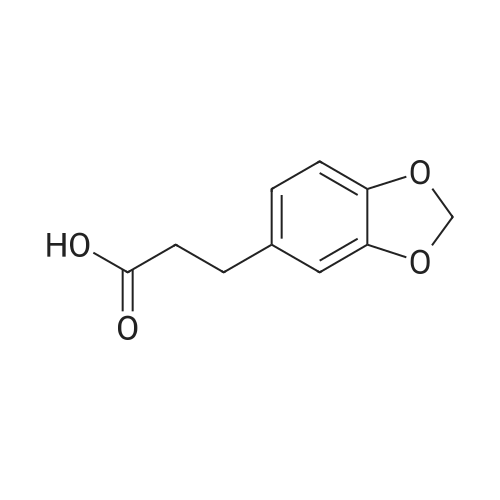
3-Benzo[1,3]dioxol-5-yl-propionic acid (2.50 g, 12.9 mmol) and thionyl chloride (20 mL, 274 mmol) were heated to 79 C. for 4 h then concentrated to afford 3-(benzo[d][1,3]dioxol-5-yl)propanoyl chloride as a brown oil. 2-(2-Imidazol-1-yl-6-methyl-pyrimidin-4-yl)-2-methyl-propylamine (100 mg, 0.432 mmol) and methylene chloride (20 mL) were added and the reaction mixture stirred at rt for about 2 h. The reaction mixture was filtered, filtrate washed with K2CO3 (100 mL, sat. aq.), and the organic layer was dried over Na2SO4. Filtration, concentration and purification using column chromatography gave 40 mg of 3-benzo[1,3]dioxol-5-yl-N-[2-(2-imidazol-1-yl-6-methyl-pyrimidin-4-yl)-2-methyl-propyl]-propionamide as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.90 (s, 1H), 7.92 (s, 1H), 7.05 (s, 1H), 6.90 (s, 1H), 6.70 (m, 3H), 5.90 (s, 2H), 3.59 (d, 2H), 2.94 (t, 2H), 2.57 (s, 3H), 2.40 (t, 2H), 1.28 (s, 6H). |
|
With pyridine; thionyl chloride; In dichloromethane; at 0 - 20℃; for 1h; |
General procedure: A solution of 1 mmol of the corresponding propanoic acid mixed with one drop of pyridine in 10 mL of dichloromethane (DCM),cooled at 0C, was added dropwise to 1.2 mmol of thionyl chloride.After stirring for 1 h at rt, the crude was evaporated at reducedpressure. The crude was dissolved in DCM at 0 C and mixed with1.2 mmol of the corresponding amine reagent and with 0.8 mmol oftriethylamine. After stirring for 3 h, the crude was diluted with 40 mL of DCM and washed with water (3 100 mL). The organicphase was separated, dried with Na2SO4 and concentrated at reduced pressure. Purification was performed by silica gelchromatography. |
|
With thionyl chloride; In toluene; for 4h;Reflux; |
General procedure: After dissolving the carboxylic acid in toluene (10 ml), SOCl2 (1 ml) was added and the reaction mixture was refluxed. The toluene was removed under vacuum and the crude mixture was dissolved in CH2Cl2 under N2 atmosphere. The alcohol and Et3N were added together to the solution and the reaction mixture was stirred at room temperature for some hours. Brine was added to the mixture and, then, it was extracted three times with CH2Cl2; the organic layers were collected, dried over Na2SO4 and the solvent was evaporated under reduced pressure. |
|
With thionyl chloride; In dichloromethane; N,N-dimethyl-formamide; at 55℃; for 5h; |
General procedure: To a solution of carboxylic acid (1 equiv.) and SOCl2 (5 equiv) inCH2Cl2 (0.5 M) was added 3 drops of DMF. The reaction was refluxed at 55 C for 5 h. ThenCH2Cl2 and the excess SOCl2 (5 equiv) were removed by vacuum. The acyl chloride obtainedwas dissolved in CH2Cl2 (1 M) for the next step. After that, to a 0 C solution of8-Aminoquinoline (10.4 mmol, 1 equiv.) and NEt3 (1.05 equiv.) in CH2Cl2 (0.5 M) was added theacyl chloride (1.05 equiv.) solution dropwise over 10 min. The solution was allowed to warm toambient temperature and was stirred for 6 h. After that, the solution was washed with water (15mL), sat. NaHCO3 (15 mL) and brine (15 mL). The organic phase was dried over Na2SO4,concentrated and purified by flash chromatography to provide the desired product. |
|
With oxalyl dichloride; In dichloromethane; for 4h;Inert atmosphere; |
To a stirred solution of carboxylic acid 30 (0.22 g,1.2 mmol) in CH2Cl2 (3 mL), under an atmosphere of nitrogen, was added oxalyl chloride (0.2 mL, 2.3mmol) dropwise and the mixture stirred for 4 h. The solvent was removed in vacuo to give the titlecompound 34b (0.24 g, quant.) as a green oil, which was placed under nitrogen and used withoutfurther purification. |
|
With oxalyl dichloride; In dichloromethane; at 20℃; for 4h; |
1,3-Benzodioxole -5-formaldehyde and malonic acid are used as starting materials, 1,3-benzodioxole -5-propionic acid is generated by simple redox reaction, corresponding amide is generated by nucleophilic substitution reaction with (S) 1,3 -benzyl -5 -2 -4-oxazolidinone under the action of n-butyl lithium, and at the same time, under the action of the ligand LiHMDS, it undergoes a nucleophilic substitution reaction with the bromopropyl group to produce the corresponding olefin, andis reduced, hydrolyzed, and oxidized under the catalysis of thecombined oxidant OsO4/NMO to produce the corresponding product, and then uses the strong reducing agent LiAlH4and the oxidizing agent NaIO4,the carbonyl compound is made into the corresponding hydroxyl compound by the two-step reaction of reduction and oxidation, and the Fetizon reagent is used to selectively oxidize it to the lactone, which is combined with 5-(bromomethyl)-1,3-benzodi The substitution reaction of catalyzer occurs under the action of LDA, and the final product is reduced with diisobutylaluminum hydride to obtain the product. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping