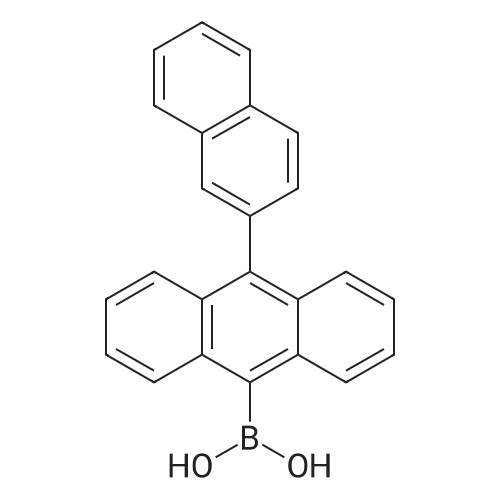
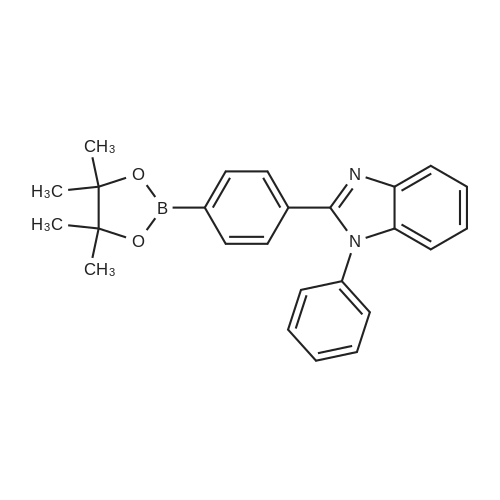
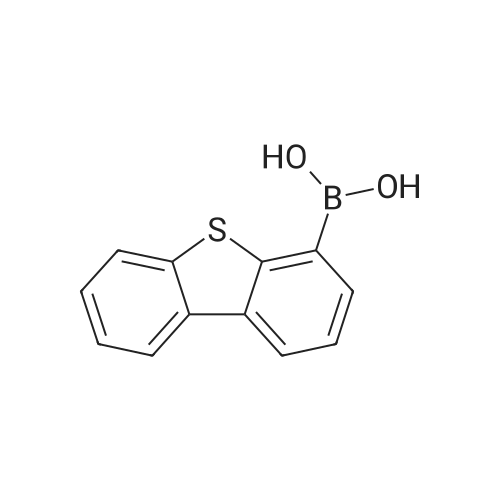
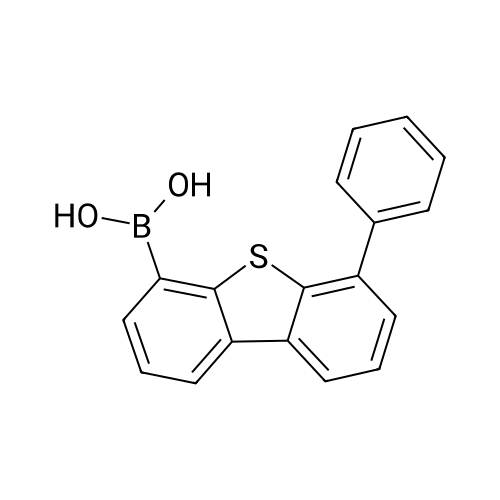
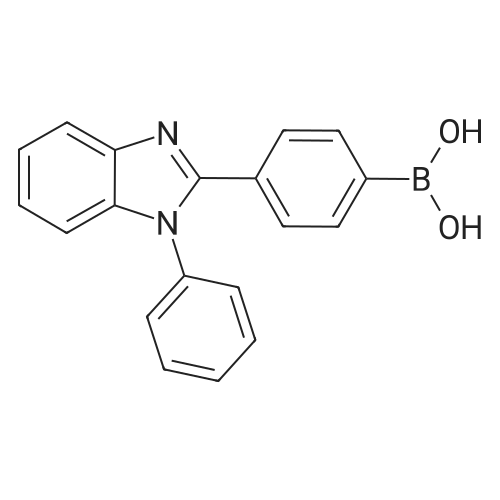
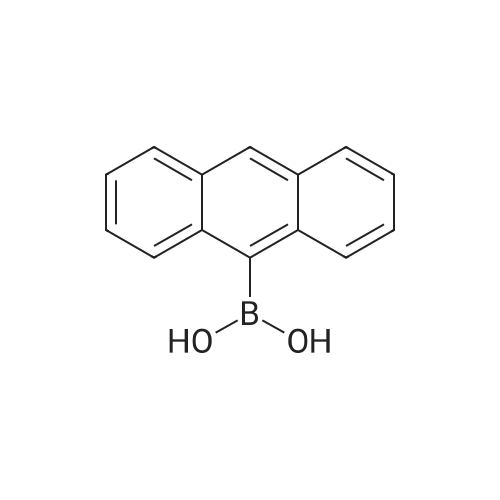
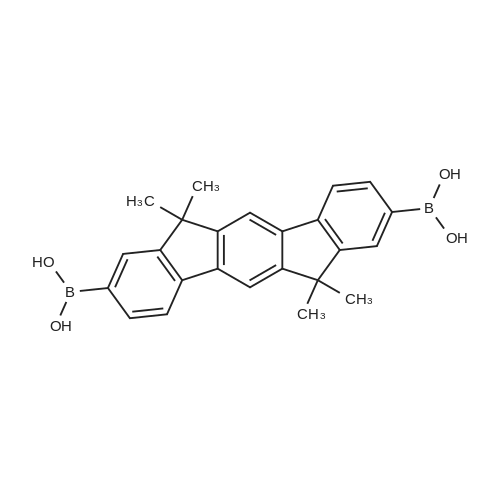
| 90% |
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In 1,4-dioxane; at 40 - 80℃;Inert atmosphere; |
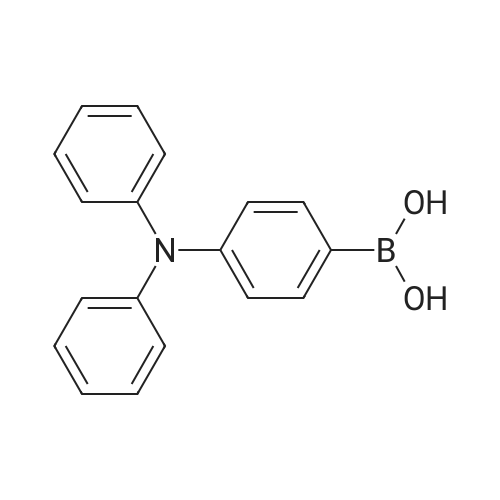
A procedure from the literature [Ge, Z.; Hayakawa, T.; Ando, S.; Ueda, M.; Akiike, T.; Miyamoto, H.; Kajita, T.; Kakimoto, M. Chem. Mater. 2008, 20(7), 2532-2537] was modified as follows: a mixture of Compound 1 (36.00 g, 103.1 mmol), bis(pinacolato)diboron (28.80 g, 113.4 mmol), [1,1′-bis(diphenylphosphino)-ferrocene]dichloropalladium(II) (3.771 g, 5.154 mmol), potassium acetate (30.35 g, 309.3 mmol) and 1,4-dioxane (480 mL) was degassed with argon for about 1 h at about 40 C. while stirring. The reaction mixture was then maintained under argon at about 80 C. while stirring. Upon confirming consumption of the starting material by TLC (SiO2, 9:1 hexanes-ethyl acetate), the reaction was cooled to RT and poured over ethyl acetate (EtOAc) (ca. 1.6 L). The mixture was then filtered through a short silica gel plug (ca. inch) and the filtrant washed copiously with EtOAc (ca. 200 mL). The combined organics were then washed with sat. NaHCO3, water and brine, dried over MgSO4, filtered and concentrated in vacuo. Purification of the crude product via flash chromatography (SiO2, 4:1 to 1:1-hexanes:ethyl acetate) afforded Compound 2 (36.8 g, 90%) as an off-white powder: confirmed by LCMS (APCI): calculated for C25H26BN2O2 (M+H+): 397; Found: 397. |
| 84.1% |
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In N,N-dimethyl-formamide; at 120 - 150℃; for 10h;Inert atmosphere; |
In a 500 mL three-necked flask,Access to nitrogen,0.05 Intermediate N1 was added to 300 ml of N, N-dimethylformamide (DMF)Then 0.06 mol bis (pinacolato) diboron,0.0005 mol of (1,1'-bis (diphenylphosphino) ferrocene) dichloropalladium (II) and0.125 mol of potassium acetate was added,Stirring the mixture,The mixed solution of the above reactants was reacted at a reaction temperature of 120-150 CUnder reflux for 10 hours;After the reaction,Cool and add 200 ml of water,And the mixture was filtered and dried in a vacuum oven.The obtained residue was separated and purified on a silica gel column,To give the compound intermediate C1;HPLC purity 99.5%, yield 84.1%. |
| 84.1% |
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In N,N-dimethyl-formamide; at 120 - 150℃; for 10h;Inert atmosphere; |
(3) in 500mL three-necked flask,Purged with nitrogen,0.05 was added as an intermediate N1 to 300 ml of N, N-dimethylformamide (DMF)0.06 mol of bis (pinacolato) diboron,0.0001 mol (1,1'-bis (diphenylphosphino) ferrocene) dichloropalladium (II) and 0.125 mol of potassium acetate were added,The mixture was stirred,The mixed solution of the above reactants was heated to reflux for 10 hours at a reaction temperature of 120-150 C;After the reaction was over, 200 ml of water was cooled and added, and the mixture was filtered and dried in a vacuum oven.The obtained residue was separated and purified on a silica gel column to obtain Compound Intermediate B1;HPLC purity 99.5%, yield 84.1%. |
| 84.1% |
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetylacetonate; In N,N-dimethyl-formamide; for 10h;Inert atmosphere; Reflux; |
(3) In a 500mL three-necked flask, purged with nitrogen, 0.05N intermediate N1 was dissolved in 300mlN, N- dimethylformamide (DMF), then 0.06mol bis (pinacolato) diboron,0.0001 mol (1,1'-bis (diphenylphosphino) ferrocene) dichloropalladium (II) and 0.125 mol of potassium acetate were added and the mixture was stirred. The mixed solution of the above reactants was stirred at a reaction temperature of 120 to 150 C Heated to reflux for 10 hours;After the reaction was over, 200 ml of water was cooled and added, and the mixture was filtered and dried in a vacuum oven.The obtained residue was separated and purified on a silica gel column to obtain Compound Intermediate B1; HPLC purity 99.5%, yield 84.1%. |
| 84% |
With tris-(dibenzylideneacetone)dipalladium(0); potassium acetate; tricyclohexylphosphine; In 1,4-dioxane; at 100℃;Inert atmosphere; |
Reaction process: tris(dibenzylideneacetone)dipalladium (1.05 g, 1.145 mmol)Tricyclohexylphosphine (0.8 g, 2.86 mmol) was added to a dry 1,4-dioxane solvent, and the air was replaced with nitrogen three times and activated at room temperature for 30 minutes.E-1 (20.0 g, 57.27 mmol) was added to the reaction mixture.Pinacol borate (17.45 g, 68.724 mmol) and potassium acetate (22.45 g, 229.08 mmol). Replace the air three times with nitrogen,The reaction solution was heated to 100 C.Treatment: TLC monitors the reaction. After the reaction is over,Cool to room temperature under nitrogen. Using diatomaceous earth to remove the catalyst,The filter cake was rinsed with dichloromethane until no product. Concentrate the filtrate to a little,The concentrated droplets were added to petroleum ether with stirring. When the solids are completely precipitated,Filtering and drying to obtain intermediate G-1(19.06 g, yield 84%). |
| 82% |
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; potassium acetate; In 1,4-dioxane; at 80℃; for 18h; |
Preparation of Compound F[186] Compound F-2 (38.6g, 110mmol), potassium acetate (32.4g, 330mmol), bis(pinacolato)diboron (36.5g, 143mmol), 1,4-dioxane (390mL), and PdCl2(dppf) (1.8g, 2mmol) were mixed and stirred at 80 for 18 hours, and then cooled at room temperature. Water (400mL) was added to the reactant and stirred. After stirring, an organic layer was extracted by adding saturated sodium chloride aqueous solution and ethyl acetate, dried over magnesium sulfate, and treated with activated charcoal, followed by filtering with celite. A solid prepared by concentrating the filtrate under reduced pressure was re-crystallized, thereby obtaining Compound F (36g, 82%).[187] 1H NMR (CDCl3) d 7.92(d, 1H), 7.76(d, 2H), 7.59(d, 2H), 7.56-7.45(m, 3H), 7.41-7.29(m, 4H), 7.27(s, 1H), 1.35(s, 12H) |
| 81% |
With potassium acetate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; at 80℃;Inert atmosphere; |
Example 1.3.3; 10; [0087] l-phenyl-2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)- lH-benzo[d] imidazole (10): A mixture of Compound 9 (0.70 g, 2 mmol), bis(pinacolate)diborane (0.533 g, 2.1 mmol), l,l'-Bis(diphenylphosphino)ferrocene] dichloropalladium (Pd(dppf)Cl2) (0.060 g, 0.08 mmol) and anhydrous potassium acetate (0.393 g, 4mmol) in 1,4-dioxane (20 mL) was heated at 800C under argon overnight. After cooling to r.t., the whole mixture was diluted with ethyl acetate (80 mL) then filtered. The solution was absorbed on silica gel, then purified by column chromatography (hexanes/ethyl acetate 5:1 to 3:1) to give a white solid (Compound 10) (0.64 g, in 81% yield). |
| 81% |
With potassium acetate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; at 80℃;Inert atmosphere; |
Example 1.1.3 1-phenyl-2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1H-benzo[d]imidazole (3): A mixture of Compound 2 (0.70 g, 2 mmol), bis(pinacolate)diborane (0.533 g, 2.1 mmol), bis(diphenylphosphino)ferrocene]dichloropalladium (Pd(dppf)Cl2) (0.060 g, 0.08 mmol) and anhydrous potassium acetate (KOAc) (0.393 g, 4 mmol) in 1,4-dioxane (20 ml) was heated at 80 C. under argon overnight. After cooling to RT, the whole was diluted with ethyl acetate (80 ml) then filtered. The solution was absorbed on silica gel, then purified by column chromatography (hexanes/ethyl acetate 5:1 to 3:1) to give a white solid 3 (0.64 g, in 81% yield). |
| 81% |
With potassium carbonate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; at 80℃;Inert atmosphere; |
Example 1.3.3 1-phenyl-2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1H-benzo[d]imidazole (10): A mixture of Compound 9 (0.70 g, 2 mmol), bis(pinacolate)diborane (0.533 g, 2.1 mmol), 1,1'-Bis(diphenylphosphino)ferrocene]dichloropalladium (Pd(dppf)Cl2) (0.060 g, 0.08 mmol) and anhydrous potassium acetate (0.393 g, 4 mmol) in 1,4-dioxane (20 mL) was heated at 80 C. under argon overnight. After cooling to r.t., the whole mixture was diluted with ethyl acetate (80 mL) then filtered. The solution was absorbed on silica gel, then purified by column chromatography (hexanes/ethyl acetate 5:1 to 3:1) to give a white solid (Compound 10) (0.64 g, in 81% yield). |
| 81% |
With potassium acetate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; at 80℃;Inert atmosphere; |
Example 1.1.3bis(pinacolate)diborane81%[0044] l-phenyl-2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)-lH- benzo[d]imidazole (3): A mixture of Compound 2 (0.70 g, 2 mmol), bis(pinacolate)diborane (0.533g, 2.1 mmol), bis(diphenylphosphino)ferrocene]dichloropalladium (Pd(dppf)Ci2) (0.060 g, 0.08 mmol) and anhydrous potassium acetate (KOAc) (0.393 g, 4mmol) in 1,4-dioxane (20 ml) was heated at 80 C under argon overnight. After cooling to RT, the whole was diluted with ethyl acetate (80 ml) then filtered. The solution was absorbed on silica gel, then purified by column chromatography (hexanes/ethyl acetate 5:1 to 3:1) to give a white solid 3 (0.64 g, in 81% yield) |
| 81% |
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In 1,4-dioxane; at 80℃; for 12h;Inert atmosphere; Schlenk technique; |
A mixture of 1a (0.70 g , 2.0 mmol), bis(pinacolate)diborane (0.53 g, 2.1 mmol), l,l'-bis(diphenylphosphino)ferrocene]dichloropalladium (Pd(dppf)Cl2) (0.060g,0.08mmol), andanhydrous potassium acetate (0.393 g, 4.0 mmol) in 1,4-dioxane(20 mL) was heated at 80C under nitrogen for 12 h. After cooling to room temperature, the mixture was extracted with ethyl acetate (30 mL*3). The organic extracts were washed with water, brine, and then dried with anhydrous MgSO4. After filtration, the filtrate was pumped dry in vacuo. The crude residue was subjected to column chromatography by eluting with hexanes/EA (8:1) as the eluent to give a white solid (yield: 81%). 1H NMR (400 MHz, acetone-d6): d (ppm ) 7.79 (d, J 7.6 Hz, 1H), 7.71 (d, J=7.6 Hz, 2H), 7.62-7.56 (m,5H), 7.45 (d, J=7.6 Hz, 2H), 7.37-7.23 (m, 3H) and 1.34 (s, 12H); 13C NMR (125 MHz, acetone-d6): d (ppm) 152.9, 144.3, 138.6, 138.2, 135.2, 134.0, 131.0, 129.7, 129.6, 128.6, 124.3, 123.7, 120.7, 111.4, 84.9, 25.3; Mass (FAB ): m/z: 397.1 [M+H]+. |
| 81% |
With palladium bis[bis(diphenylphosphino)ferrocene] dichloride; potassium acetate; In 1,4-dioxane; at 80℃;Inert atmosphere; |
[122] 1 -phenyl-2-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)phenyl)-1 H-benzo[d]imidazole (5): A mixture of Compound 4 (0.70 g, 2 mmol), bis(pinacolate)diborane (0.533g, 2.1 mmol), bis(diphenylphosphino)ferrocene]dichloropalladium (Pd(dppf)CI2) (0.060 g, 0.08 mmol) and anhydrous potassium acetate (KOAc) (0.393 g, 4mmol) in 1 ,4-dioxane (20 ml) was heated at about 80 C under argon overnight. After cooling to RT, the whole was diluted with ethyl acetate (80 ml) then filtered. The solution was absorbed on silica gel, then purified by column chromatography (hexanes/ethyl acetate 5:1 to 3: 1 ) to give a white solid 5 (0.64 g, in 81 % yield). |
| 81% |
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In 1,4-dioxane; at 80℃;Inert atmosphere; |
A mixture of Compound 2 (0.70 g, 2 mmol), bis(pinacolate)diborane (0.533 g, 2.1 mmol), bis(diphenylphosphino)ferrocene]dichloropalladium (Pd(dppf)C12) (0.060 g,0.08 mmol) and anhydrous potassium acetate (KOAc) (0.3 93g, 4 mmol) in 1 ,4-dioxane (20 ml) was heated at 80 C. under argon overnight. Afier cooling to RT, the whole was diluted with ethyl acetate (80 ml) then filtered. The solution wasabsorbed on silica gel, then purified by column chromatog-raphy (hexanes/ethyl acetate 5:1 to 3:1) to give a white solid3 (0.64 g, in 81% yield). |
| 81% |
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In 1,4-dioxane; at 80℃;Inert atmosphere; |
Example 1.1.3 1-phenyl-2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1H-benzo[d]imidazole (Compound 3) A mixture of Compound 2 (0.70 g, 2 mmol), bis(pinacolate)diborane (0.533 g, 2.1 mmol), bis(diphenylphosphino)ferrocene]dichloropalladium (Pd(dppf)Cl2) (0.060 g, 0.08 mmol) and anhydrous potassium acetate (KOAc) (0.393 g, 4 mmol) in 1,4-dioxane (20 ml) was heated at about 80 C. under argon overnight. After cooling to RT, the whole was diluted with ethyl acetate (80 mL) then filtered. The solution was absorbed on silica gel, then purified by column chromatography (hexanes/ethyl acetate 5:1 to 3:1) to give a white solid 3 (0.64 g, in 81% yield). |
| 81.2% |
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In N,N-dimethyl-formamide; at 120 - 150℃; for 10h; |
(3) In a 500 mL three-necked flask, nitrogen gas was introduced, and 0.05 mol of the intermediate Y1 was added and dissolved in 300 ml of N,N-dimethylformamide (DMF).Further, 0.06 mol of bis(pinacolyl)diboron, 0.0005 mol of (1,1'-bis(diphenylphosphino)ferrocene)dichloropalladium (II), and 0.125 mol of potassium acetate were added, and the mixture was stirred.Mixing the above reactants at the reaction temperatureHeating under reflux at 120-150 C for 10 hours;After the reaction was completed, it was cooled and 200 ml of water was added, and the mixture was filtered and dried in a vacuum oven.The obtained residue was separated and purified through silica gel column to give Compound Intermediate B1; HPLC purity 99.2%, yield: 81.2%. |
| 79% |
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In 1,4-dioxane; at 85℃; for 20h;Inert atmosphere; |
General procedure: In a 250 mLreaction flask, the intermediate IV (2.00 g, 3.78 mmol), bis(pinacolato)diboron (1.44 g, 5.67 mmol), potassium acetate (1.49 g, 15.18 mmol)and Pd (dppf)Cl2 (0.17 g, 0.23 mmol) were dissolved in dry 1,4-dioxane(80 mL) and refluxed for 20 h under a nitrogen atmosphere. Aftercooling to room temperature, the mixture was poured into the distilledwater and then extracted with dichloromethane. The organic phase wasdried over anhydrous Na2SO4 and concentrated in vacuum. The crudeproduct was purified by silica gel column chromatography using petroleum ether/dichloromethane (2:1, v/v) as eluent to obtain a whitesolid (1.52 g, 69.8 |
| 78% |
With bis-triphenylphosphine-palladium(II) chloride; potassium acetate; In tetrahydrofuran; at 80℃; for 3h;Inert atmosphere; |
Compound 2 (2.8g, 8.02mmol), diboron pinacol ester (3.1g, 12.03mmol) and potassium acetate (2.36g, 24.06mmol) were added to a certain amount of tetrahydrofuran (80ml), In the N2 atmosphere, Add bistriphenylphosphorus palladium dichloride (168mg, 0.24mmol), The reaction was stirred at 80C for 3 hours. After the reaction is over, Distill under reduced pressure to remove tetrahydrofuran, Dissolve with dichloromethane, Add distilled water and extract with dichloromethane. The obtained organic layer was dried with anhydrous magnesium sulfate and filtered, Distilled under reduced pressure to remove dichloromethane, The crude product obtained is separated by column chromatography, The eluent is a mixed solvent of dichloromethane and ethyl acetate (3:1v/v), A white solid (Compound 3) was obtained with a yield of 78% (2.5 g). |
|
With potassium acetate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; at 80℃;Inert atmosphere; |
[0076] l-phenyl-2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)- lH-benzo[d]imidazole (3): A mixture of compound (2) (0.70 g, 2 mmol), bis(pinacolate)diborane (0.533g, 2.1 mmol), Pd(dppf)Cl2 (0.060 g, 0.08 mmol) and anhydrous potassium acetate (0.393 g, 4mmol) in 1,4-dioxane (20 mL) was heated at about 80 C under argon overnight. After cooling to room temperature, the whole was diluted with ethyl acetate (~80 mL) then filtered. The solution was absorbed on silica gel, then purified by column chromatography (hexanes/ethyl acetate 5: 1 to 3:1) to give a white solid (0.64 g, in 81% yield). |
|
|
Under N2 gas purification system, Compound A, 1.5 equivalents of Bu-Li into diethyl ether, and the mixture was stirred at a temperature of -78 deg. C. After 2 hours the reaction was complete, added 1.2 equivalents of compound B, and the mixture was stirred at a temperature of -78 deg. C for 30 minutes. The dry ice bath was removed and the reaction temperature was raised to room temperature. After 8 hours the reaction was complete, deionized water, dilute HCl (30ml) to remove the organic solvent. After complete removal of the organic solvent, water and the precipitated white solid was filtered off to obtain a compound C. |
|
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In 1,4-dioxane; at 80℃;Glovebox; |
In a glovebox, three oven-dried reaction jars were each charged with 2-(4- bromophenyl)-l -phenyl- lH-benzimidazole 12 (4.20 g, 12.0 mmol), bis(pinacolato)- diboron (Bpin) (3.05 g, 12 mmol, 1 eq.), potassium acetate (2.95 g, 30 mmol, 2.5 eq.), and Pd(dppf)Cl2 (300 mg, 0.36 mmol, 3 mol%). The solvent (1,4-dio-xane, 80 mL) was added to each jar and the reactions were stirred at 80 C overnight. After complete conversion (-95%) as determined by LC-MS, the contents of the reaction jars were mixed and the solvent removed under reduced pressure. Water (200 mL) was added the product was extracted into chloroform (3x200 mL), the product was passed through a silica plug and recrystallized from acetonitrile. The reaction was successfully scaled up to 12 g in 90% yield after recrystallization to give the desired boronic ester 8 at -98% purity as judged by LC-MS and NMR. *H NMR (500 MHz, cdcl3) δ 7.90 (dt, J = 8.1, 0.9 Hz, 1H), 7.76 - 7.71 (m, 2H), 7.60 - 7.55 (m, 2H), 7.49 - 7.41 (m, 3H), 7.35 - 7.21 (m, 6H), 1.32 (s, 12H); 13C NMR (126 MHz, CDC13) δ 172.37, 157.10, 152.56, 135.25,133.58, 133.11, 131.25, 130.46, 128.49, 128.13, 128.03, 127.96, 127.73, 127.11, 126.47, 125.24, 124.99, 123.30 |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping