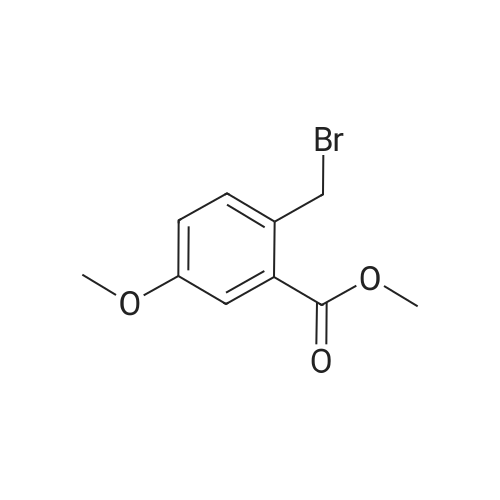
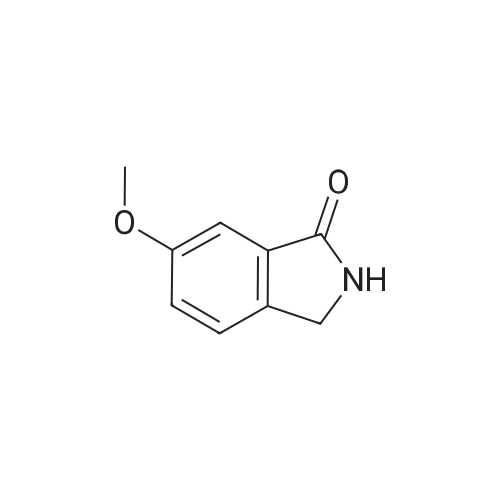
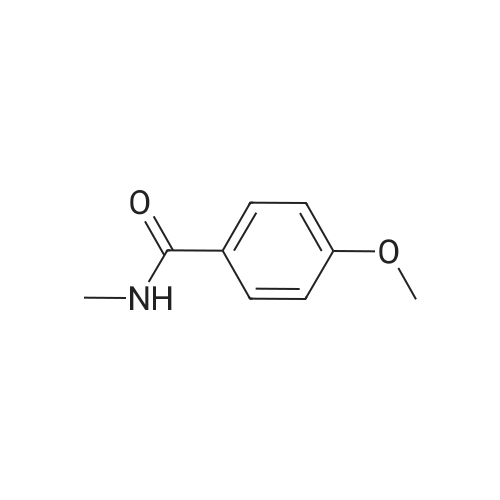
| 43% |
With ammonium cerium(IV) nitrate; water; In acetonitrile; at 20℃; for 1h; |
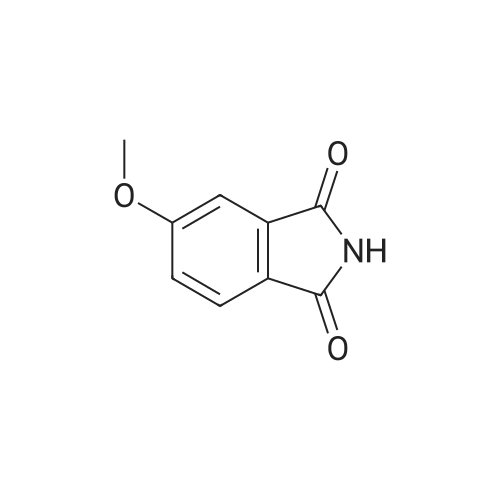
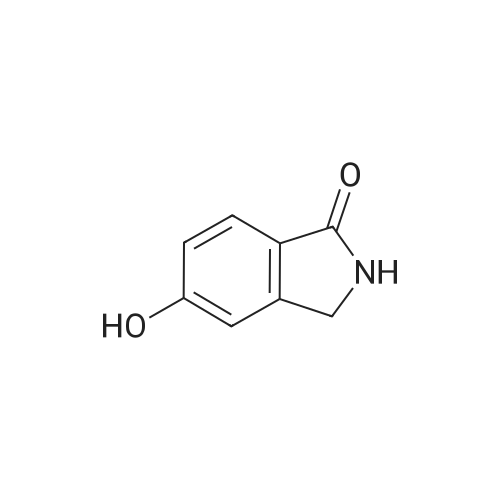
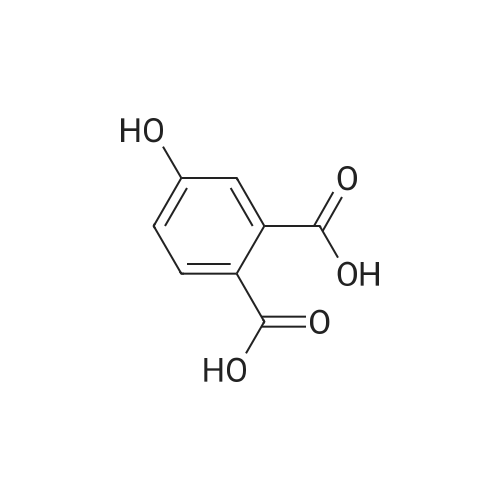
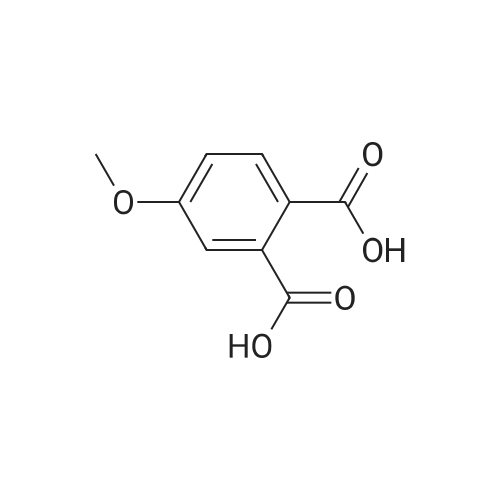
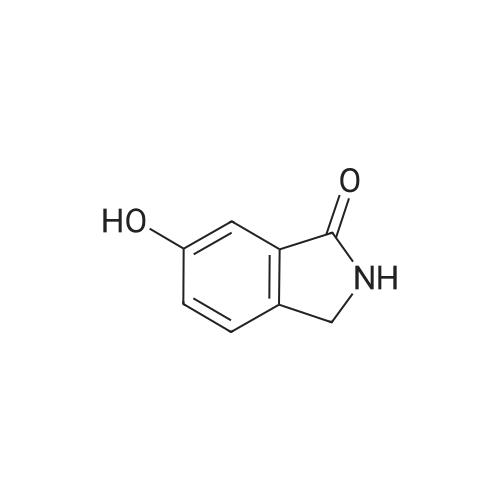
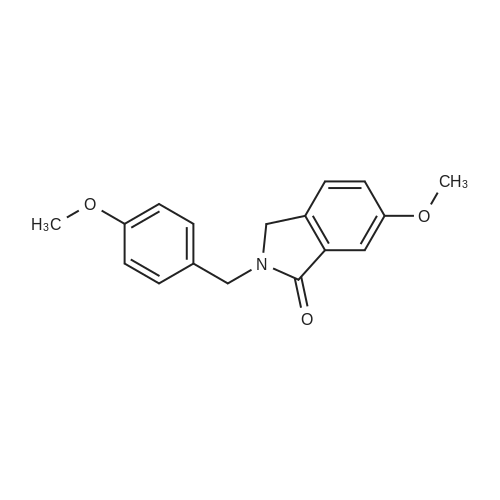
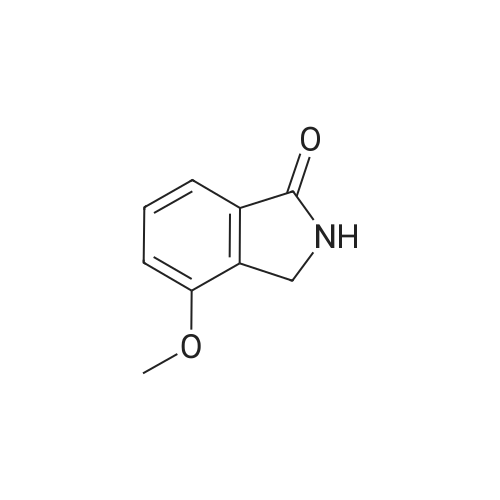
A mixture [OF 6-METHOXY-2- (4-METHOXY-BENZYL)-2, 3-DIHYDRO-ISOINDOL-1-ONE] (300 mg, 1.1 mmol) and ammonium ceric nitrate (2.2 g, 4.0 mmol) in acetonitrile: water (12 mL, 2: 1) was stirred at RT for 1 h. Then the mixture was poured into water and extracted with ethyl acetate. The combined extracts were then washed with sodium hydrogen carbonate and water. The organic layer was then dried over sodium sulphate, filtered and evaporated. The residue was then purified by chromatography [(SI02,] dichloromethane: [MEOH] 20: 1) to afford the title product (75 mg, 43%) as a light yellow solid. MS m/e = 164.2 (M). Alternatively, a mixture [OF 6-METHOXY-2- (4-METHOXY-BENZYL)-2, 3-DIHYDRO-ISOINDOL-1-ONE] (98.5 mg, 0.35 mmol) in dichlormethane (10 mL) containing TFA (1.6 mL, 20.8 mmol) and TfOH (0.6 mL, 7.0 mmol) was heated overnight at [40 C.] Then the mixture was poured into sodium hydrogen carbonate and water and the product extracted with di- chloromethane. The combined organic layers were then dried over sodium sulphate, filtered and evaporated. The residue was then purified by chromatography [(SI02,] dichloro- methane: [MEOH] 20: 1) to afford the title product (24 mg, 42%) as a light yellow solid. MS m/e = 164.2 (M). i) 6-Hydroxy-2, [3-DIHYDRO-ISOINDOL-1-ONE] A mixture [OF 6-METHOXY-2, 3-DIHYDRO-ISOINDOL-1-ONE] (167 mg, 1.0 mmol) and boron tri- bromide (1 M in [CH2C12,] 3.6 mL, 3.6 mmol) in [CH2CI2] (8 mL) [AT-78C] was stirred for 18 h at RT. The mixture was then cooled to-78C and [MEOH] (20 mL) was added. After 2 h at-78C the mixture was evaporated and the residue purified by chromatography [(SI02,] [CH2CL2] : 2N NH3-MeOH 9: 1) to afford the title product (147 mg, 100%) as a white solid. MS m/e = 148.0 (M-H+). |
| 42% |
With trifluorormethanesulfonic acid; trifluoroacetic acid; In dichloromethane; at 40℃; |
A mixture [OF 6-METHOXY-2- (4-METHOXY-BENZYL)-2, 3-DIHYDRO-ISOINDOL-1-ONE] (300 mg, 1.1 mmol) and ammonium ceric nitrate (2.2 g, 4.0 mmol) in acetonitrile: water (12 mL, 2: 1) was stirred at RT for 1 h. Then the mixture was poured into water and extracted with ethyl acetate. The combined extracts were then washed with sodium hydrogen carbonate and water. The organic layer was then dried over sodium sulphate, filtered and evaporated. The residue was then purified by chromatography [(SI02,] dichloromethane: [MEOH] 20: 1) to afford the title product (75 mg, 43%) as a light yellow solid. MS m/e = 164.2 (M). Alternatively, a mixture [OF 6-METHOXY-2- (4-METHOXY-BENZYL)-2, 3-DIHYDRO-ISOINDOL-1-ONE] (98.5 mg, 0.35 mmol) in dichlormethane (10 mL) containing TFA (1.6 mL, 20.8 mmol) and TfOH (0.6 mL, 7.0 mmol) was heated overnight at [40 C.] Then the mixture was poured into sodium hydrogen carbonate and water and the product extracted with di- chloromethane. The combined organic layers were then dried over sodium sulphate, filtered and evaporated. The residue was then purified by chromatography [(SI02,] dichloro- methane: [MEOH] 20: 1) to afford the title product (24 mg, 42%) as a light yellow solid. MS m/e = 164.2 (M). i) 6-Hydroxy-2, [3-DIHYDRO-ISOINDOL-1-ONE] A mixture [OF 6-METHOXY-2, 3-DIHYDRO-ISOINDOL-1-ONE] (167 mg, 1.0 mmol) and boron tri- bromide (1 M in [CH2C12,] 3.6 mL, 3.6 mmol) in [CH2CI2] (8 mL) [AT-78C] was stirred for 18 h at RT. The mixture was then cooled to-78C and [MEOH] (20 mL) was added. After 2 h at-78C the mixture was evaporated and the residue purified by chromatography [(SI02,] [CH2CL2] : 2N NH3-MeOH 9: 1) to afford the title product (147 mg, 100%) as a white solid. MS m/e = 148.0 (M-H+). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping