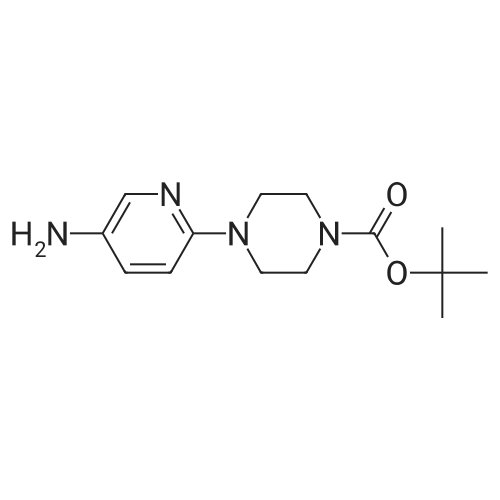
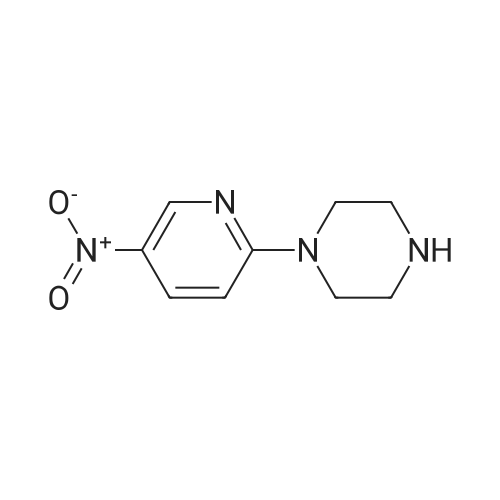
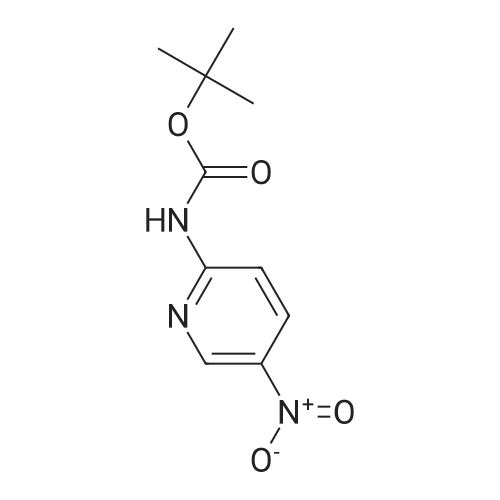
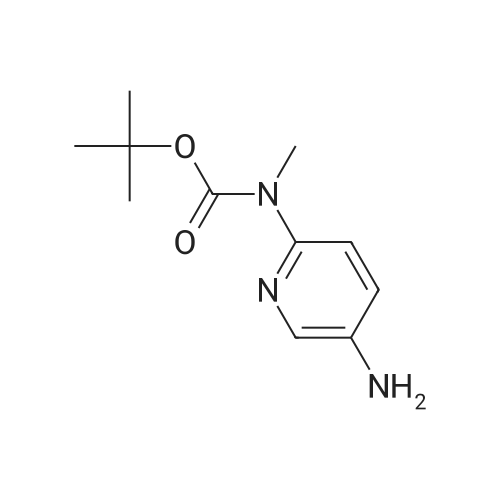
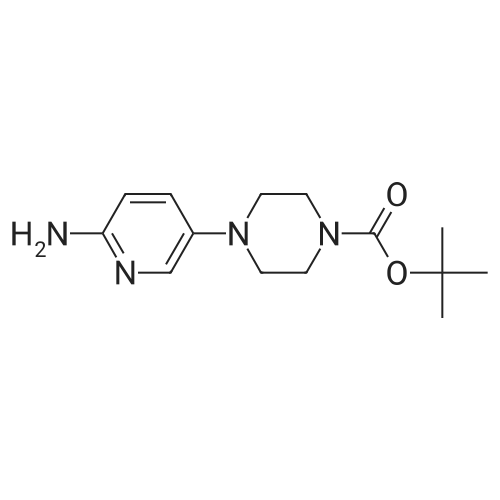
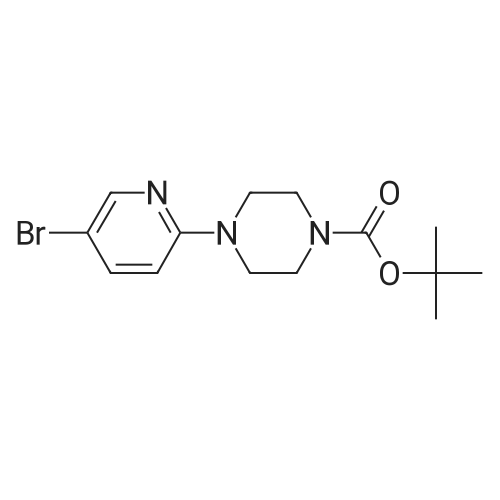
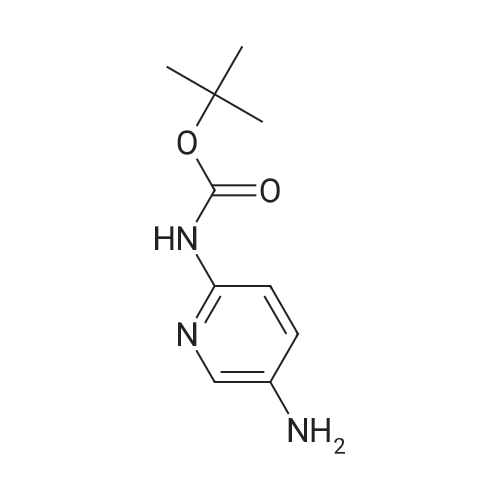
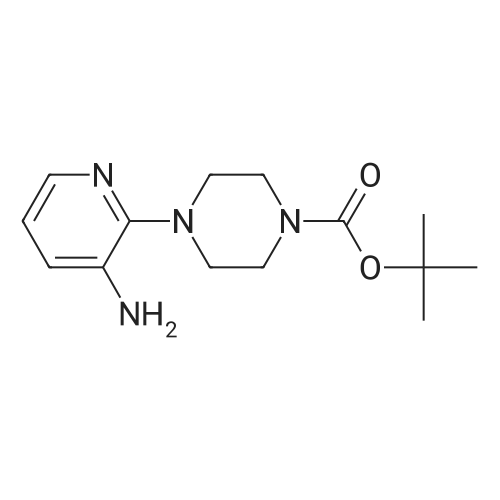
| 95% |
With potassium carbonate; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 120℃; for 4h; |
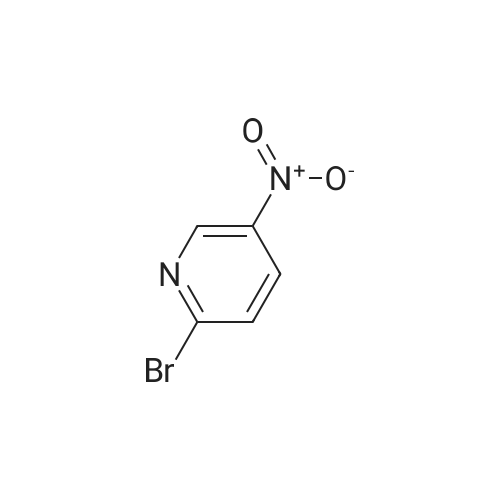
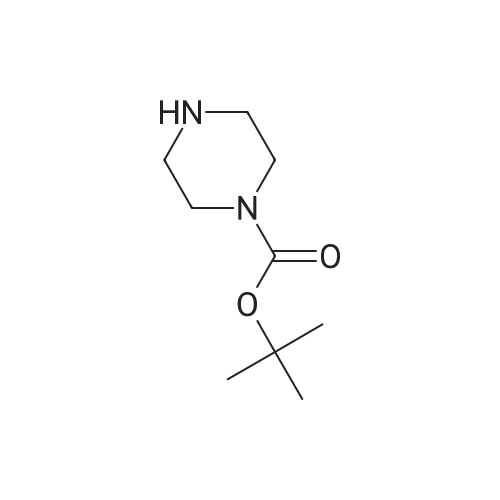
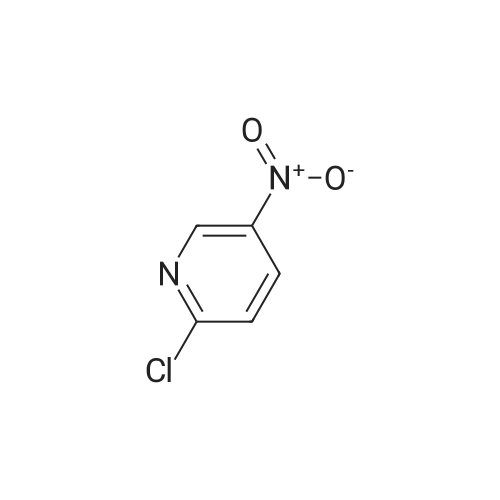
N-BOC piperazine (0.5g) and 2-chloro-5-nitropyridine (0.424g) in DMF (16ml) were treated with potassium carbonate (0.744g) and DIPEA (1.41ml). The resulting mixture was heated at 1200C for 4 hours.After cooling to room temperarure, the solvent was removed in vacuo and the residue partitioned between ethyl acetate and water. Organic layer washed again with water then dried over anhydrous magnesium sulphate. After filtration, the solvent evaporated to dryness in vacuo to afford the title compound as a pale brown solid in 95%. LCMS (ES+) 209.05 (MH+-BOC). |
| 90% |
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 70℃; for 5h; |
To a stirred solution of compound 46-1 (2-Chloro-5-nitro-pyridine) (1 g, 6.30 mmol) inAcetonitrile (20 mL), Piperazine-1-carboxylic acid tert-butyl ester 46-2 (1.17 g, 6.30 mmol) andDIPEA (3.29 mL, 18.92 mmol) were added and the reaction was heated at 70 oc for 5 hours. Thereaction mixture was diluted with ethyl acetate and washed with water and brine, dried over sodiumsulfate and concentrated to afford compound 46-3 (1.75 g, 90%) as yellow solid. LC MS: ES+ 309.0. |
| 88% |
With potassium carbonate; In 1,4-dioxane; for 4h;Heating / reflux; |
Potassium carbonate (1.7g, 12.31 mmol) was added to a solution of 2-Chloro- 5-nitropyridine (1.33g,8.38mmol) and piperazine-1-carboxylic acid tert-butyl ester (1.57g, 8.42mmol) in dioxane (10ml) then stirred at reflux for 4hours. The reaction was cooled, and solvent evaporated. The residue was extracted with MeCI2 (100ml) washed with H2O (50ml), separated organic layer washed with brine (50ml) dried over MgSO4, filtered and solvent evaporated yielding a residue which chromatographed on silica gel eluting with 10% v/v EtOAc/hexanes yielding desired product as a pale yellow solid (2.3g,88%)Partial 1H NMR (400MHz, CDCI3)? 9.0 (s,1H) 8.20(d,1H)6.50 (d,1 H) . |
| 87% |
With potassium carbonate; In acetonitrile; for 4h;Heating / reflux; |
A suspension of 2-chloro-5-nitropyridine (7.90 g), piperazine-1-carboxylic acid tert-butyl ester (11.2 g) and potassium carbonate (6.90 g) in acetonitrile (250 mL) was heated under reflux for four hours. The reaction mixture was concentrated and diluted with ethyl acetate and washed with water and saturated brine and dried over sodium sulfate and filtered and concentrated. The residue was vigorously stirred in ethyl acetate/isopropyl ether, collected by filtration and dried under reduced pressure, and 16.1 g (87%) of the title compound was obtained as a yellow solid. MS(FAB) m/z:309 (M + H)+. |
| 83% |
|
To a solution of tert-butyl piperazine-1-carboxylate (64 g, 346 mmol) in 600 ml. of THF at O0C was added NaH (16.4 g, 409 mmol, 60% in mineral oil) portionwise. The reaction mixture was stirred for 15 min and 2-chloro-5-nitropyridine (50 g, 314 mmol) was added. The reaction mixture was allowed to warm to rt and then heated to 5O0C for 4 h. The reaction was quenched by water (30 ml.) and extracted with DCM (1.5 Lx 3). The combined organic layers were dried over Na2SO4 and the solvent was removed under reduced pressure. The residue was subjected to wash with petroleum ether to give the desired product of Step A (80 g, yield 83%). 1H NMR (CDCI3, 400 MHz) delta 8.95 (d, J = 2.4 Hz, 1 H), 8.24 (d, J = 12 Hz, 1 H), 6.92 (d, J = 6.0 Hz, 1 H), 3.75 (s, 4 H), 3.44 (s, 4H), and 1.41 (s, 9 H). |
| 58% |
With potassium carbonate; In butan-1-ol; at 120℃; for 17h; |
Place 2-chloro-5-nitro-pyridine (1.0 g, 6.31 mmol), piperazine-1-carboxylic acid tert-butyl ester (1.76 g, 9.45 mmol), and triethylamine (1.76 mL, 12.6 mmol) in ra-butanol (20 mL). Heat to 120 C for 17 hours. Cool to room temperature and add ethyl acetate and water. Separate organic layer and wash with water and saturated aq. sodium chloride. Collect organic layer, dry over Mg2SO4, filter, and concentrate under reduced pressure. Subject residue to silica gel chromatography eluting with 20% EtOAc:hexane to yield 1.14 g (58%) of 4-(5-nitro-pyridin-2-yl)-piperazine-l-carboxylic acid tert-butyl ester. MS(ES): m/z = 309 [M+H]. |
| 50% |
|
To a solution containing 5.8 g (31.5 mmol) of tert-butyl 1-piperazinecarboxylate and 20 mL of THF at 00C was added 1.5 g (37 mmol) of a 60% dispersion of NaH in mineral oil. The reaction mixture was allowed to stir for 20 min and 5.0 g (31.5 mmol) of 2-chloro-5-nitropyridine was added. The reaction mixture was heated at 500C <n="96"/>overnight, quenched by the addition of water, and extracted with DCM. The combined organic layers were dried over MgSO4 and the solvent was removed under reduced pressure. The residue was subjected to silica gel chromatography to give 4.89 g (50%) of tert-butyl 4-(5-nitro-2-pyridinyl)-1-piperazinecarboxylate as a yellow solid: 1H NMR (400 MHz, DMSOd6) delta 8.25 (dd, J = 9.5 and 2.9 Hz, 1 H), 6.93 (d, J = 9.5 Hz, 1 H), 3.76 -3.78 (m, 4 H), 3.41 - 3.48 (m, 4 H), and 1.42 (s, 9 H). |
| 50% |
|
To a solution containing 5 8 g (31 5 mmol) of fert-butyl 1-piotaperaziotanecarboxylate and 20 mL of THF at 0 C was added 1 5 g (37 mmol) of a 60% dispersion of NaH in mineral oil The reaction mixture was allowed to stir for 20 mm and 5 0 g (31 5 mmol) of 2-chloro-5-niotatropyriotadiotane was added The reaction mixture was heated at 50 C overnight, quenched by the addition of H2O, and extracted with DCM The combined organic layers were dried over MgSO4 and the solvent was removed under reduced pressure The residue was subjected to silica gel chromatography to give 4 89 g (50%) of fert-butyl 4-(5-niotatro-2-pyriotadiotanyl)-1-piotaperaziotanecarboxylate as a yellow solid 1H-NMR (400 MHz, DMSO-d6) delta 8 25 (dd, J =9 5 and 2 9 Hz, 1 H), 6 93 (d, J =9 5 Hz, 1 H), 3 76 -3 78 (m, 4 H), 341 - 3 48 (m, 4 H), and 1 42 (s, 9 H) |
|
With copper diacetate; potassium carbonate; In water; N,N-dimethyl-formamide; at 85℃; for 3h; |
Potassium carbonate (5 g) and Cu(OAc)2.2H2O (0.4 g) were added to a solution of piperazine-1-carboxylic acid tert-butyl ester (5 g) and 2-Chloro-5-nitropyridine (4.5 g) in DMF (75 ml) and the reaction mixture was stirred at 85 C for 3 h. On completion, the reaction mixture was allowed to cool to RT and the DMF was removed under reduced pressure. Water (200 ml) was added to the reaction mixture resulting in the formation of a solid that was filtered and dissolved in ethyl acetate (400 ml). The organic layer was washed with water (200 ml) and dried over sodium sulfate. The solvent was removed under reduced pressure to give 4-(5-Nitro-pyridin-2-yl)-piperazine-l-carboxylic acid tert-butyl ester. |
|
With potassium hydrogencarbonate; In N,N-dimethyl-formamide; at 70℃; for 3h; |
Step 1 : fert-Butyl 4-(5-nitropyridin-2-yl)piperazine-l-carboxylate: To a stirred solution of 2-chloro-5-nitropyridine (20.00 g, 126.151 mmol) in DMF (200 ml) was added JV- BOC-piperazine (25.81 g, '138.766 mmol) and KHCO3 (18.94 g, 189.226 mmol) and the mixture was stirred at 70 C for 3 h. The mixture was cooled to room temperature and diluted with ethyl acetate (300 ml) and water (3 x 200 ml). The layers were separated. The aqueous layer was extracted with ethyl acetate (300 ml). The combined organic extracts were washed <n="28"/>with water (3 x 200 ml), brine (200 ml) and dried over anhydrous Na2SO4. The residue obtained after evaporation of the solvent was triturated with /-pentane to give 32.34 g of the product as a yellow solid; IR (KBr) 3430, 2975, 1691, 1512, 1292, 937 cm"1; 1H NMR (CDCl3, 300 MHz) delta 1.49 (s, 9H), 3.57 (br s, 4H), 3.77 (br s, 4H), 6.57 (d, J= 9.0 Hz, IH), 8.24 (d, J= 6.0 Hz, IH), 9.04 (s, IH); MS (ESI) m/z 413.22 (MH)+. |
|
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 100℃; for 16h; |
Intermediate 2Step A: t-butyl 4-(5-nitropyridin-2-yl)piperazine- -carboxylateTo 5-nitro-2-chloropyridine (10.0 g, 0.0631 mol) and N-BOC-piperazine (17.6 g, 0.0946 mol) dissolved in DMF (200 mL) was added N,N-diisopropylethylamine (24.5 g, 31.3 mL, 0.189 mol). The reaction mixture was heated at 100 C for 16 h then cooled to RT and concentrated. Water (300 mL) was added, and the aqueous solution was extracted with 0??. The combined organic extract was dried (MgS04), filtered, and concentrated. Purification by vacuum filtration through silica gel (eluant: 5% EtOAc/C?Cl2) to yield t-butyl 4-(5-nitropyridin-2-yl)piperazine- 1 -carboxylate as a yellow solid. LC/MS = 309 [M+l ]. |
| 4.94 g |
In ethanol; at 70℃; for 16h; |
To a solution of 2-chloro-5-nitropyridine (LXV) (2.0 g, 12.6 mmol) in EtOH (20 mL) was added tert-butyl piperazine-l-carboxylate (XCVI) (7.05 g, 37.9 mmol). The reaction was headed at 70C for 16 h. The reaction was concentrated under vacuum and then dissolved in EtOAc. The EtOAc was washed with 1 M NaOH, brine and then dried over MgS04 to give tert-butyl 4-(5-nitropyridin-2-yl)piperazine-l- carboxylate (XCVII) as a yellow solid (4.94 g). ESIMS found for C14H20N4O4 mlz 309.0 (M+H). |
| 4.94 g |
In ethanol; at 70℃; for 16h; |
Step 1 To a solution of 2-chloro-5-nitropyridine (LVII) (2.0 g, 12.6 mmol) in EtOH (20 mL) was added tert-butyl piperazine-1-carboxylate (LXXII) (7.05 g, 37.9 mmol). The reaction was headed at 70 C. for 16 h. The reaction was concentrated under vacuum and then dissolved in EtOAc. The EtOAc was washed with 1 M NaOH, brine and then dried over MgSO4 to give tert-butyl 4-(5-nitropyridin-2-yl)piperazine-1-carboxylate (LXXIII) as a yellow solid (4.94 g). ESIMS found for C14H20N4O4 m/z 309.0 (M+H). |
| 4.94 g |
In ethanol; at 70℃; for 16h; |
To a solution of 2-chloro-5-nitropyridine (LXV) (2.0 g, 12.6 mmol) in EtOH (20 mL) was added fert-butyl piperazine-l-carboxylate (XCVI) (7.05 g, 37.9 mmol). The reaction was headed at 70C for 16 h. The reaction was concentrated under vacuum and then dissolved in EtOAc. The EtOAc was washed with 1 M NaOH, brine and then dried over MgS04 to give tert-butyl 4-(5-nitropyridin-2-yl)piperazine-l-carboxylate (XCVII) as a yellow solid (4.94 g). ESIMS found for C14H20N4O4 mlz 309.0 (M+H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +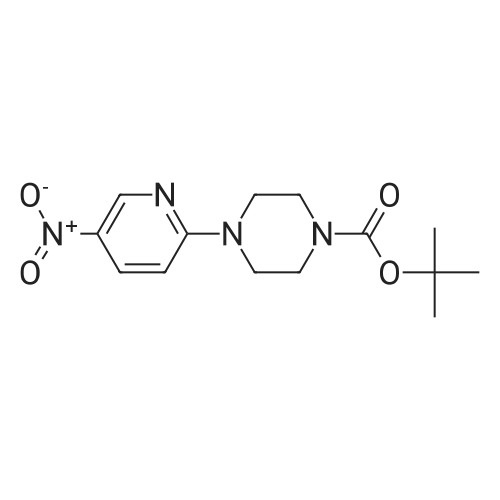

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping