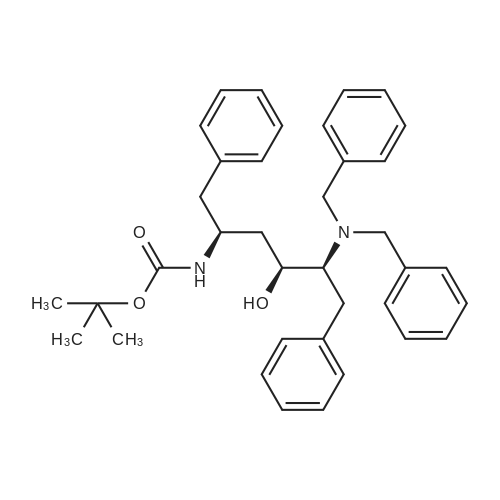
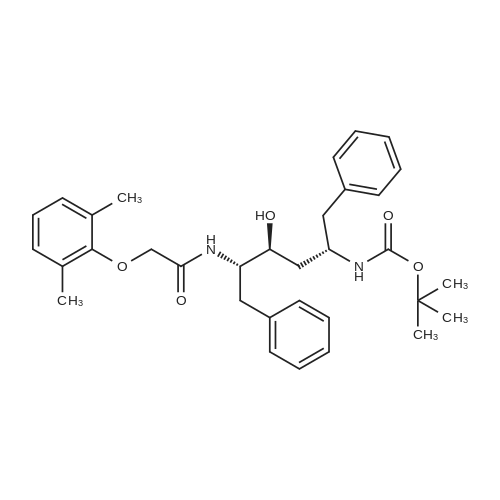
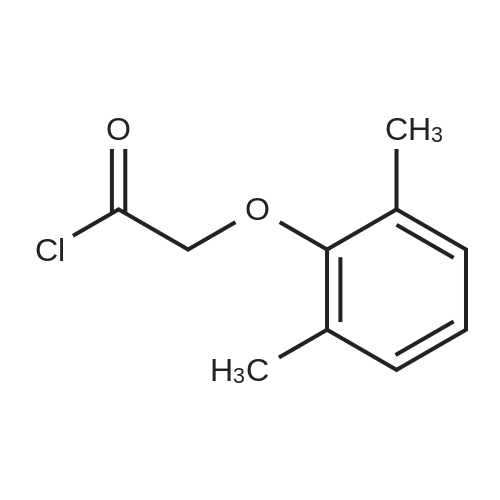
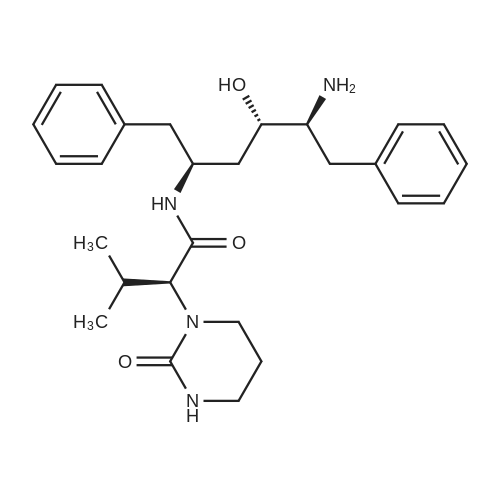
Abstract: Cryptococcosis is a fungal infection that is becoming increasingly prevalent worldwide, particularly among individuals with compromised immune systems, such as HIV patients. Amphotericin B (AmB) is the first-line treatment mainly combined with flucytosine. The scarcity and the prohibitive cost of this regimen urge the use of fluconazole as an alternative, leading to increased rates of treatment failure and relapses. Therefore, there is a critical need for efficient and cost-effective therapy to enhance the efficacy of AmB. In this study, we evaluated the efficacy of the HIV protease inhibitors (PIs) to synergize the activity of AmB in the treatment of cryptococcosis. Five PIs (ritonavir, atazanavir, saquinavir, lopinavir, and nelfinavir) were found to synergistically potentiate the killing activity of AmB against Cryptococcus strains with ?FICI ranging between 0.09 and 0.5 against 20 clinical isolates. This synergistic activity was further confirmed in a time-kill assay, where different AmB/PIs combinations exhibited fungicidal activity within 24 hrs. Additionally, PIs in combination with AmB exhibited an extended post-antifungal effect on treated cryptococcal cells for approximately 10 hrs compared to 4 hours with AmB alone. This promising activity against cryptococcal cells did not exhibit increased cytotoxicity towards treated kidney cells, ruling out the risk of drug combination-induced nephrotoxicity. Finally, we evaluated the efficacy of AmB/PIs combinations in the Caenorhabditis elegans model of cryptococcosis, where these combinations significantly reduced the fungal burden of the treated nematodes by approximately 2.44 Log10 CFU (92.4%) compared to the untreated worms and 1.40 Log10 ((39.4%) compared to AmB alone. The cost-effectiveness and accessibility of PIs in resource-limited geographical areas compared to other antifungal agents, such as flucytosine, make them an appealing choice for combination therapy.
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
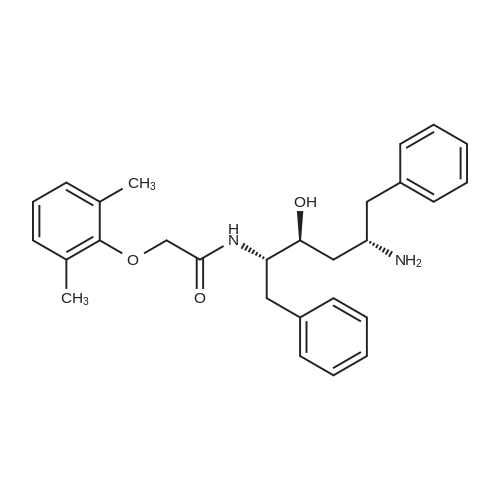
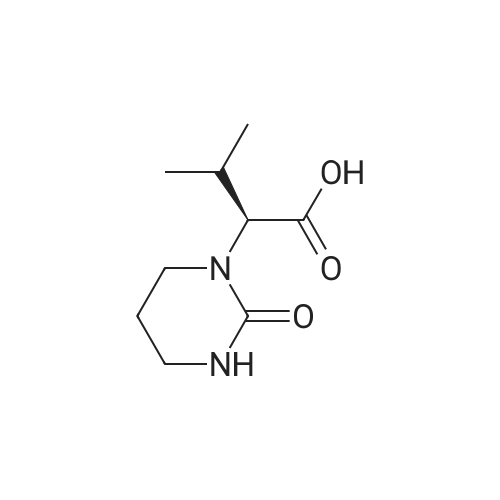
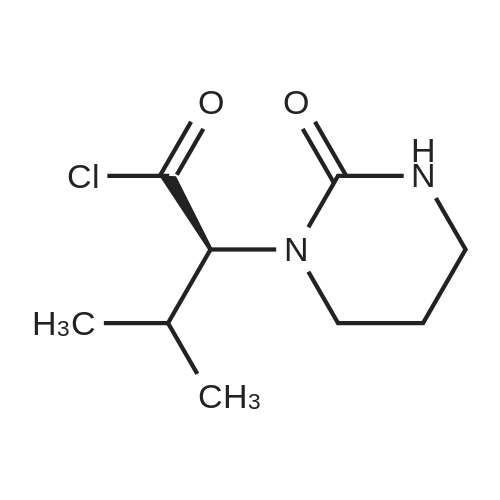
20.0 g (0.1 mol) of (2S)-(1-tetrahydropyrimidin-2-one)-3-methylbutyric acid and 100 ml of dichloromethane were added to the reaction flask.The mixture was placed under ice water, and the temperature was controlled below 10 C. 13.9 g (0.11 mol) of thionyl chloride was dropped into the reaction solution, and the addition was completed. The reaction was stirred at 0 to 10 C for 1 h.Then refluxing for 1 h to obtain (2S)-(1-tetrahydropyrimidin-2-one)-3-methylbutyryl chloride reaction solution;The reaction solution was lowered to 0-20 C, 25.3 g (0.25 mol) of triethylamine was added, and placed under ice water, and the temperature was controlled below 10 C.N-[(1S,2S,4S)-4-Amino-2-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-2-(2,6-dimethylphenoxy) Acetamide42.4g (0.095mol) was added to the reaction solution, and the addition was completed. The reaction was stirred at 0~10 C for 1 h.Then react at room temperature for 4 h,The lopinavir reaction solution was obtained; 10% sodium hydrogencarbonate 50 g was added to the lopinavir reaction solution, stirred for 1 hour, and layered.Then, the reaction liquid was washed with 25% sodium chloride 50 g and 50 g of purified water, and the layers were separated, and concentrated under reduced pressure to give an oily substance, 300 ml of ethyl acetate and 300 ml of n-heptane were added, and the mixture was heated to reflux and cooled to T=20~ At 25 C, and stir for 1 hour.Then cooled to 10 C, and stirred for 2 hours, filtered, filter cake vacuum drying at 50 C for 12 hours, 53.7 g of lopinavir finished product, yield 90.1%, HPLC purity ? 99.5%.
Example 1Thionyl chloride (18 ml) was added to the mixture of 2S-(1-tetrahydro- pyrimid-2-onyl)-3-methylbutanoic acid (25 gm), tetrahydrofuran (370 ml) and dimethylformamide (2 ml) at 0 - 10 deg C and the mass was stirred for 1 hour 15 minutes. The mass was subjected to distillation under reduced pressure to remove excess thionyl chloride, n-heptane (45 ml) was added to the residue obtained and the solvent was distilled off. The reaction mass was slurried in dimethylformamide (105 ml). (2S,3S,5S)-2-(2,6-dimethylphenoxyacetyl)amino-3- hydroxy-5-amino-1 ,6-diphenylhexane (41 gm), imidazole (25 gm) and 4- (dimethylamino)pyridine (1.5 gm) were dissolved in ethyl acetate (420 ml). To the solution was added above slurried product at 0 - 10 deg C. The reaction mass was maintained for 14 hours and then ethyl acetate (165 ml) and water (250 ml) were added. The layers were separated, water (250 ml) was added to the organic layer and the pH was adjusted to 2.0 - 3.0 with dilute hydrochloric acid (6N HCI). The layers were separated, the organic layer was washed with aqueous sodium bicarbonate and then with water. The ethyl acetate was distilled off from the mass. The reaction mass was dissolved in ethyl acetate (80 ml) and n-heptane (80 ml) was added to the solution. The separated solid was stirred with ethyl acetate (290 ml) for 8 hours, filtered and dried the solid to obtain 33 gm of lopinavir ethyl acetate solvate.
(2S,3S,5S)-2-amino-3-hydroxy-5-(2S-(1-tetrahydro-pyrimid-2-onyl)-3-methylbutanoyl)amino-1,6-diphenylhexane (S)-pyroglutamic acid salt[ No CAS ]
[ 20143-48-0 ]
[ 192725-17-0 ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydrogencarbonate; In water; ethyl acetate; at 20 - 25℃; for 1.41667h;Product distribution / selectivity;
The product obtained in Stage-3 (50 g, 0.084 mol) was added to a mixture of ethyl acetate (375 ml) and DM water (375 ml). Sodium bicarbonate (41.22 g, 0.49 mol) was added and stirred for 15 min at 20-25 C. Thereafter, the solution of acid chloride prepared in step-1 of Stage-4 was added in 10 min while maintaining a vigorous stirring at 20-25 C. After stirring for 1 h at 20-25 C., the organic layer was separated and washed with 5% aq sodium bicarbonate (250 ml) followed by 10% aq sodium chloride solution (250 ml). The organic layer was treated with activated carbon (5 g) for 15 min. The carbon was filtered off through hyflo and washed with ethyl acetate (50 ml). The combined filtrate was concentrated at 50-55 C. under reduced pressure to dryness and the resulting residue was dissolved in ethyl acetate (250 ml) and taken to dryness. The residue obtained was dissolved in ethyl acetate (250 ml) at 50-60 C. and added heptanes (250 ml). The reaction mass was stirred at 50-55 C. for an additional 1 h and the resulting slurry was cooled to room temperature. After stirring the crystallized product for 5 h at room temperature the product was isolated by filtration and washed with a mixture of ethyl acetate (50 ml) and heptanes (50 ml). The wet product was dried under reduced pressure at 50 C. to obtain 45 g of Lopinavir.
90 g
With sodium hydrogencarbonate; In water; ethyl acetate; at 25 - 35℃; for 1h;
2,6-dimethylphenoxyacetic acid (28.7 g), ethyl acetate (100 mL), thionyl chloride (25 g) and dimethyl formamide (1 mL) were charged in reaction flask at 25-35C. Reaction mass was heated to 50-55C and stirred for 2-3 hrs at same temperature, and then added to a mixture of pyroglutamic acid salt of Formula VI (100 g), ethyl acetate (750 mL), water (750 mL) and sodium bicarbonate (82.5 g) at 25-35C and stir for 60 min at same temperature. After completion of the reaction, organic layer was separated and treated with 5% sodium bicarbonate solution (25 g dissolved in 500 mL). Then the organic layer was separated and concentrated under vacuum at below 60C to get the title compound as a residue. Yield: 100 g; HPLC purity: 99%; Formula D by HPLC: 0.15%; Formula E by HPLC: Not detected; and Formula F by HPLC: 0.8%. (0179) The above lopinavir (100 g) was dissolved in methanol (300 mL) and ammonia (100 mL) was charged and the reaction mass was heated to 40-45C for 1-2 hrs at same temperature. After completion of the reaction, solvent was concentrated under vacuum at below 60C to obtain residue. To the obtained residue methylene chloride (400 mL) and water (200 mL) were charged at 25-35C. Then the product containing organic layer was speared and concentrated under vacuum at below 45C. The obtained solid was dissolved in acetone (200 mL) at 50-55C for 30-60 min. Reaction mass was gradually cool to 0- 5C and the precipitated solid was filtered and washed with chilled acetone (100 mL) and dried at 30-35C under vacuum to get the title compound. Yield: 90 g; HPLC purity: 99.9%; Formula A by HPLC: Not detected; Formula B by HPLC: Not detected; Formula C by HPLC: Not detected; Formula D by HPLC: 0.02%; Formula E by HPLC: Not detected; and Formula F by HPLC: Not detected.
With dibenzoyl peroxide; In acetonitrile; at 0℃; for 3h;Product distribution / selectivity;
Example 6A; r2SVN-l(lS.3S,4SVl-benzvl-4-[[(2.6-dimethvlphenoxv')acetvl1ammol-3-[l-Cethylthio)ethoxy]-5-phenylpentyl) -3-methyl-2-(2-oxotetrahydropyrimidin-1 (2HVvDbutanamide; Method A; To a solution of the compound of Example 3 (0.50 g, 0.80 mmol) and ethyl sulfide(2.1 mL) in acetonitrile (6 mL) at 0C was added benzoyl peroxide (1.16 g) in three portionsover 3 hours. The reaction was diluted with ethyl acetate and washed with 10% NaaCOs andbrine. The organic was dried over MgSO/i, filtered and evaporated. The residue waschromatographed on silica gel eluting with a gradient of 50-100% ethyl acetate in chloroformto give the title compound (0.36 g, 61% yield).
MethodB; A slurry of N-chlorosuccinimide (6.5 g, 48.7 mmol) in tetrahydrofuran (50 mL) wascooled to -10C, followed by addition of diethyl sulfide (7.0 mL) and then by addition of asolution of Example 3 (5.0 g) in tetrahydrofuran (20 mL). A solution of triethylamine (9.0mL) in tetrahydrofuran (15 mL) was then added dropwise and the mixture was stirred at -10C for 1.5 h. The reaction was quenched with 10% Na2CO3 and extracted twice with ethylacetate. The combined organic was washed with water and brine and dried over MgSCU,filtered and evaporated to give the crude product (7.5 g).
With acetic anhydride; acetic acid; at 20℃; for 48 - 72h;Product distribution / selectivity;
Example 4A; (2SVN-((lS,3S,4SVl-benzvl-4-fr(2.6-dimetfavlphenoxv)acetvl]ainino)-3-r(methvltMo)methoxy]-5-phenvlpentvl>-3-methyl-2-(2-oxotetrahydropvrimi(Hn-l(2HVyDbutanamide; Method A;To a solution of the compound of Example 3 (3.0 g, 4.8 mmol), DMSO (18 mL), andacetic acid (3.6 mL) at room temperature was added acetic anhydride (23 mL), and thereaction was stirred for 48 hours at room temperature. The reaction was quenched with iceand 10% NaaCOs was added to adjust the pH to 7. The mixture was extracted with ethylacetate and washed with 10% NaaCOs and brine. The organic was dried over Na2SO4,filtered and evaporated to give the crude product, which was chromatographed on silica geleluting first with 25-100% ethyl acetate in dichloromethane to give the title compound (2.1 g,64% yield).; Method B; Example 3 (50.4 g, 0.080 mol), 85 mLDMSO (15 equivalents), 75mL aceticanhydride (10 equivalents), 135 ml acetic acid (30 equivalents) were mixed at ambienttemperature under nitrogen for 3 days. The reaction was quenched with 1500 mL aqueous17% Na2COs pre-chilled to 0C. The mixture was extracted with 1400 mL ethyl acetate andthen with 500 mL ethyl acetate twice. The organic layers were combined and washed with700 mL 10% Na2CO3, water 600 mL x3, and 500 ml saturated brine, sequentially. Theorganic layer was dried over MgSC>4, concentrated and chased with heptanes to give 56.4 g oftitle compound as white foam.
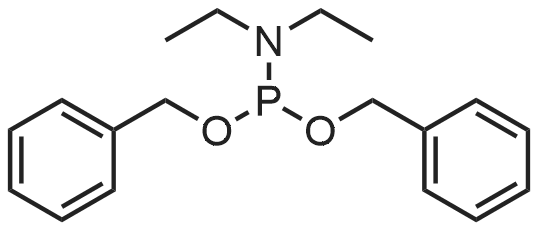
dibenzyl (1S,3S)-1-((1S)-1-[(2,6-dimethylphenoxy)acetyl]amino}-2-phenylethyl)-3-[(2S)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanoyl]amino}-4-phenylbutyl phosphate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
Example 7A; dibenzvl (1S 3S>-1 -((1 S)-1 - [(2.6-dimethylphenoxy)acetyl] amino} -2-phenylethyl)-3 - [(2S)-3 -methyl-2-(2-oxotetrahydropyrimidin-1 (2HV vDbutanoyl] amino} -4-phenylbutyl phosphate; A solution of the compound of Example 3 (0.250 g, 0.40 mmol), dibenzyldiethylphosphoramidite (0.28 mL), and IH-tetrazole (0.14 g) in THF (4.0 mL) was stirred atroom temperature for 68 hours. Dichloromethane (4.0 mL) was added and the mixture wascooled to -45C, followed by addition of m-chloroperbenzoic acid (0.089 g). After stirringfor 30 minutes at -45C, the reaction was diluted with ethyl acetate and washed twice with10% Na2CO3 and then with brine. The organic phase was dried over MgSC>4, filtered andevaporated. The residue was chromatography on silica gel, eluting with a gradient startingwith 33% ethyl acetate in chloroform and ending with 5% methanol in ethyl acetate, to givethe title compound (0.324 g, 90% yield).
(1S,3S)-1-((1S)-1-[(2,6-dimethylphenoxy)acetyl]amino}-2-phenylethyl)3-[(2S)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanoyl]amino}-4-phenylbutyl 4-[bis(benzyloxy)phosphoryl]oxy}-3,3-dimethylbutanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84%
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 20℃; for 19h;
Examle 9D; 2-(2-oxotetrahydropvrirmdin-l (2HVyl)butanovl]amino} -4-phenylbutyl 4-[bisfbenzyloxv^phosphoryljoxyl-S^-dimethylbutanoate; To a solution containing the compound of Example 3 (0.075 g, 0.12 mmol), theproduct from Example 9C (0.056 g), and DMAP (0.017 g) in DMF (1.2 mL) was addedEDAC (0.027 g) and the mixture was stirred at room temperature for 16 hours. Additionalproduct from Example 9C (0.056 g) and EDAC (0.027 g) were added and the reaction wasstirred at room temperature for 3 hours. The solvent was evaporated and the reaction mixturewas partitioned between ethyl acetate and aqueous NaHCOs. The organic layer was washedwith brine, dried over MgSO/i, filtered and evaporated. The residue was purified bychromatography on silica gel, eluting with a gradient starting with 33% ethyl acetate inchloroform and ending with 5% methanol in chloroform, to give the title compound (0.101 g,84%).
(1S,3S)-1-((1S)-1-[(2,6-dimethylphenoxy)acetyl]amino}-2-phenylethyl)-3-[(2S)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanoyl]amino}-4-phenylbutyl 4-[bis(benzyloxy)phosphoryl]oxy}-2,2-dimethylbutanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
56%
With dmap; In dichloromethane; at 0 - 20℃; for 16h;
Example 1; IDq6'3^4-rri^-l-(r(2.6-dimethvlphenoxv)acetvl]amino)-2-phenvlethvlV3-(r('2^-3-methvl-2-r2-oxotetrahvdropyrimidin-1 (2ff)-yl)butanoyl] amino > -4-phenylbutyl 4-irbis(benzyloxy')phosphorvl1oxv>-2,2-dimethvlbutanoate; To a solution containing the product from Example 11C (0.538 mmol) dissolved indichloromethane (1.0 mL) at 0C, were added the product of Example 3 (0.085 g, 0.134mmol) and 4-(dimethylamino)pyridine (0.066 g) and the reaction was allowed to warm toroom temperature and was stirred for 16 hours. The reaction was diluted withdichloromethane, and the organic layer was washed with 10% citric acid, water and brine,dried over MgSO4, filtered and evaporated. The residue was purified by chromatography onsilica gel, eluting with a gradient starting with dichloromethane and ending with 5% methanolin ethyl acetate, to give the title compound (0.075 g, 56%).
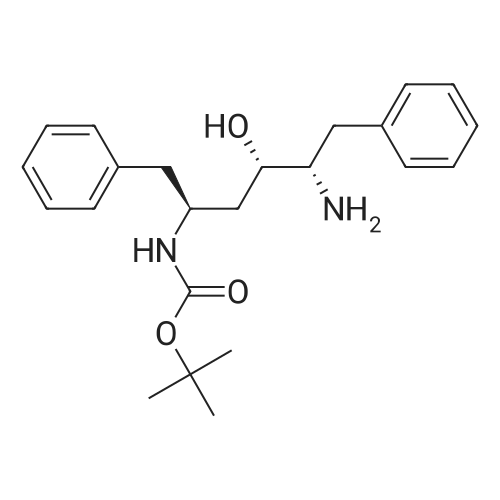
To a stirred solution of (2S,3S,5S)-2-(2,6-dimethylphenoxyacetyl)amino-3- hydroxy-5-(t-butyloxycarbonylamino)-l,6-diphenylhexane (5.1 g) in methylene chloride (50 rnL) was added trifluoroacetic acid (10.65 g) at room temperature over a period of 20 minutes. The reaction mixture was stirred at room temperature for 3 hours. The solvent was removed under reduced pressure and then water (100 mL) was added followed by methylene chloride (100 mL). To the cooled at about 10-15C biphasic mixture, solid sodium bicarbonate (12 g) was added portionwise, and finally its pH was adjusted to about 8.5 with aqueous sodium hydroxide. The organic layer was separated and washed with water (50 mL). Concentration of methylene chloride layer provided a residue as thick oil. Ethyl acetate (50 mL) was added into the above residue and stirred to yield a clear solution. To the stirred solution imidazole (2.1 g) was added at room temperature. The mixture was cooled to 00C. To the cold reaction mixture a suspension of acid chloride in dimethyl formamide (from step 1 of Example 5) was slowly added at about 0 to 50C during 30 minutes. The reaction mixture was stirred at 0 to 50C for next 30 minutes, then warmed to room temperature and stirred at room temperature for 12 hours. The reaction mixture was cooled down to 10C and quenched with aqueous hydrochloric acid (100 mL) at 10-150C. Ethyl acetate (50 mL) was added to the mixture and stirred at room temperature for 30 minutes. The organic layer was separated and washed with aqueous sodium bicarbonate (50 mL) followed by water (2 X 50 mL). Evaporation of the solvent under reduced pressure afforded crude material as an off-white solid which was dissolved in ethyl acetate (28 mL) at 45-5O0C and then heptane (28 mL) was slowly added at 50- 450C. The resulting clear solution was slowly allowed to cooled to room temperature and stirred at room temperature for 12 hours. A white solid, precipitated out from the solution was filtered and washed with 1:1 mixture of ethyl acetate and heptane (5 mL). The solid was dried under vacuum at 50-60C for 12 hours to yield the title compound as a white solid.Yield: 3.5 g
EXAMPLE 9A Preparation of Crude Lopinavir Crude lopinavir, prepared according to U.S. Pat. No. 5,914,332 (Example 38) from (2S,3S,5S)-2-amino-3-hydroxy-5-[2S-(1-tetrahydropyrimid-2-onyl)-3-methylbutanoyl]amino-1,6-diphenylhexane (S)-pyroglutamic acid salt (about 85 g, corrected for solvent content), was dissolved in 318.5 grams of ethyl acetate and the solution was concentrated to an oil in vacuo. The residue was dissolved in 225 grams of ethyl acetate, then concentrated to an oil in vacuo twice. The residue was dissolved in ethyl acetate (approximately 300 mL) at 65 C., filtered to remove any trace undissolved solids, and concentrated to a foam in vacuo. The foam was dissolved in 338 grams of ethyl acetate and this solution was divided into four equal portions.
18
[ 192725-49-8 ]
[ 192725-17-0 ]
Yield
Reaction Conditions
Operation in experiment
Step 2:; (2S,3S,5S)-2-(2,6-dimethylphenoxyacetyI)amino-3-hydroxy-5-[2s-(l- tetrahydro-pyrimid-2-onyl)-3-methylbutanoyl]amino-l,6-diphenylhexaneImidazole (2.1 g) in ethyl acetate (50 ml) was added to a stirred solution of N-[(1S,2S,4S)- 4-amino-l-benzyl-2-hydroxy-5-phenylpentyl]-2-(2,6-dimethylphenoxy)acetamide (4.2 g), at room temperature (20-250C). The mixture was cooled to 00C. To the cold reaction mixture, the suspension of acid chloride in dimethyl formamide (obtained from Step 1 of Example 3) was slowly added at 0 to 5C during 30 minutes. The reaction mixture was stirred at 0 to 50C for next 30 minutes, and then warmed to room temperature (20-250C) and stirred at room temperature (20-250C) for 12 hours. The reaction mixture was again EPO <DP n="12"/>cooled to 1O0C and quenched with aqueous hydrochloric acid (100 ml, 0.2N) at 10-15C. Ethyl acetate (50 ml) was added into the mixture and stirred at room temperature (20 to 25C) for 30 minutes. The layers were separated and the organic layer was washed with aqueous sodium bicarbonate (50 ml, 5% w/v) followed by washing with water (2 X 50 ml). The solvent was evaporated under reduced pressure to get crude material as an off- white solid. The solid so obtained was dissolved in ethyl acetate (28 ml) at 45-500C and then heptane (28 ml) was slowly added at 50-450C. The resulting clear solution was slowly allowed to cool to room temperature and stirred at room temperature for 12 hours. A white solid, precipitated out from the solution was filtered and washed with 1 : 1 mixture of ethyl acetate and heptane (5 ml). It was dried under vacuum at 50-600C for 12 hours to get the title compound as a white solid.Yield: 3.5 g
(2S,3S,5S)-2-amino-3-hydroxy-5-(2S-(1-tetrahydropyramid-2-only)-2-methylbutanoyl)-amino-1,6-diphenylhexane[ No CAS ]
[ 20143-48-0 ]
[ 192725-17-0 ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydrogencarbonate; In water; ethyl acetate; at 20 - 25℃; for 1.16667h;
The product obtained in Stage-3 (50 g, 0.084 mol) was added to a mixture of ethyl acetate (375 ml) and DM water (375 ml). Sodium bicarbonate (41 .22 g, 0.49 mol) was added and stirred for 15 min at 20-250C. Thereafter, the solution of acid chloride prepared in step- 1 of Stage-4 was added in 10 min while maintaining a vigorous stirring at 20-250C. After stirring for I h at 20-250C, the organic layer was separated and washed with 5% aq sodium bicarbonate (250 ml) followed by 10% aq sodium chloride solution (250 ml). The organic layer was treated with activated carbon (5 g) for 15 min. The carbon was filtered off through hyflo and washed with ethyl acetate (50 ml). The combined filtrate was concentrated at 5O-55C under reduced pressure to dryness and the resulting residue was dissolved in ethyl acetate (250 ml) and taken to dryness. The residue obtained was dissolved in ethyl acetate (250 ml) at 50-600C and added heptanes (250 ml). The reaction mass was stirred at 5O-55C for an additional 1 h and the resulting slurry was cooled to room temperature. After stirring the crystallized product for 5 h at room temperature the product was isolated by filtration and washed with a mixture of ethyl acetate (50 ml) and heptanes (50 ml). The wet product was dried under reduced pressure at 5O0C to obtain 45 g of Lopinavir. Purity of Lopinavir: 99.5% by HPLC






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping