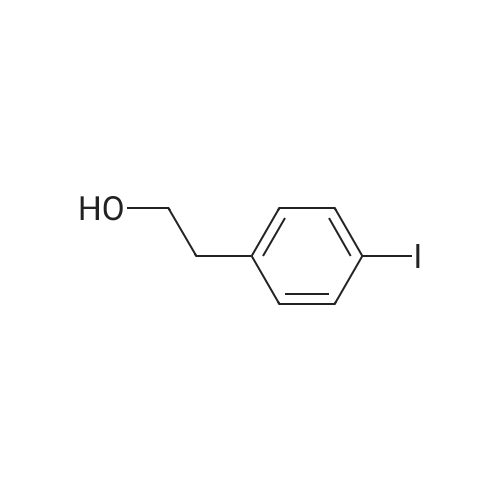
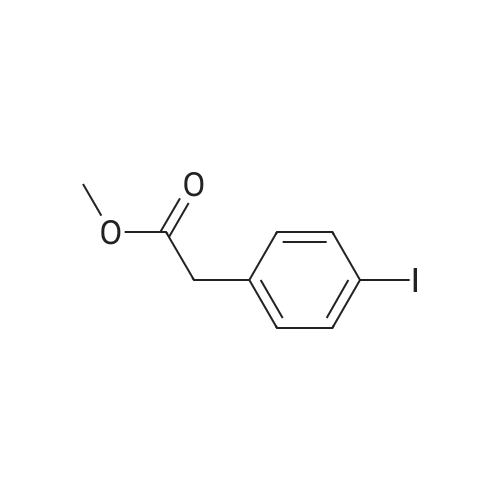
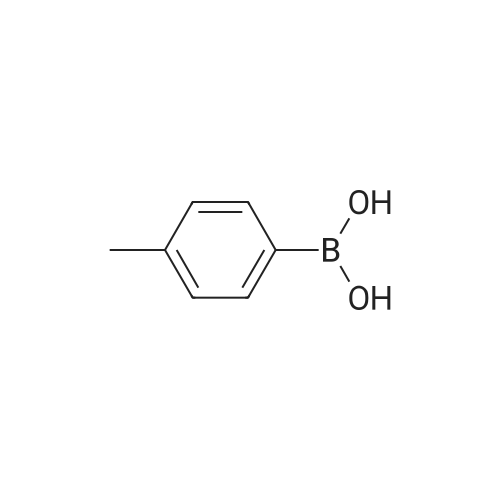
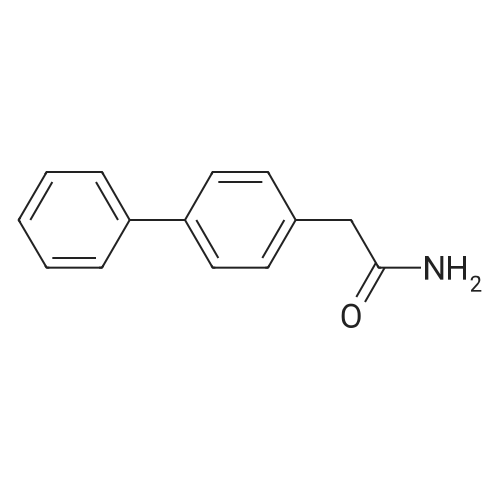
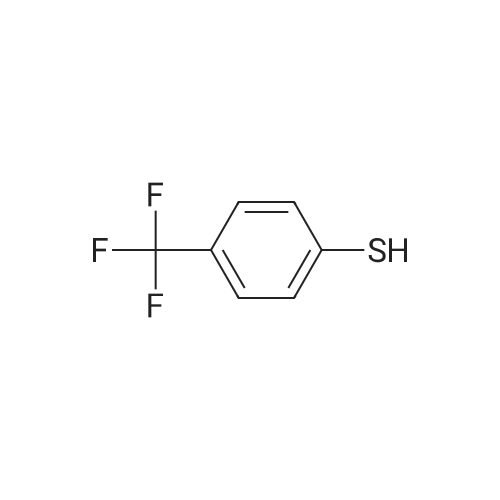
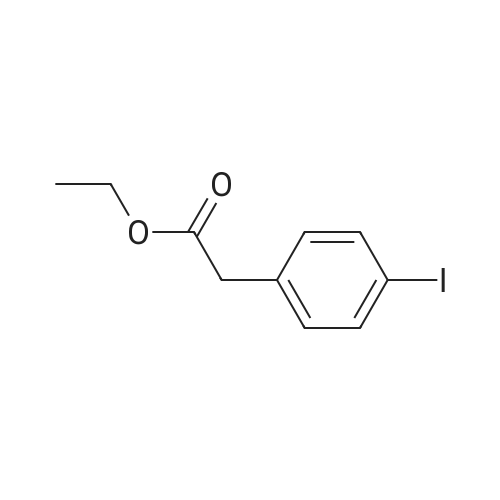
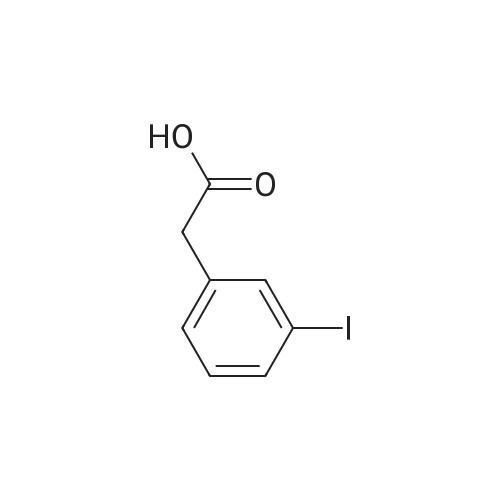
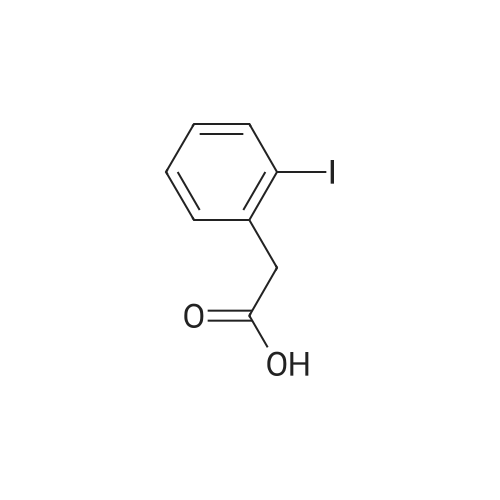
| 98% |
hydrogenchloride; In 1,4-dioxane; at 20℃; for 24h; |
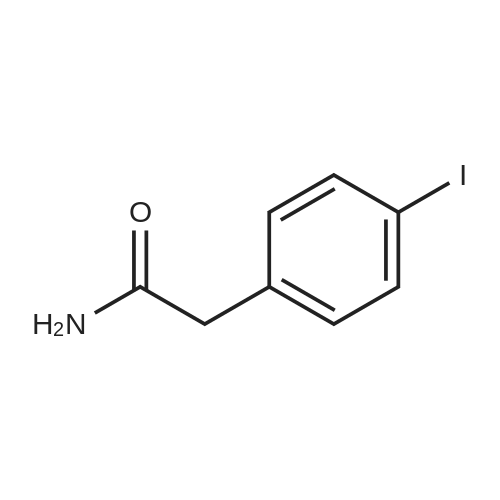
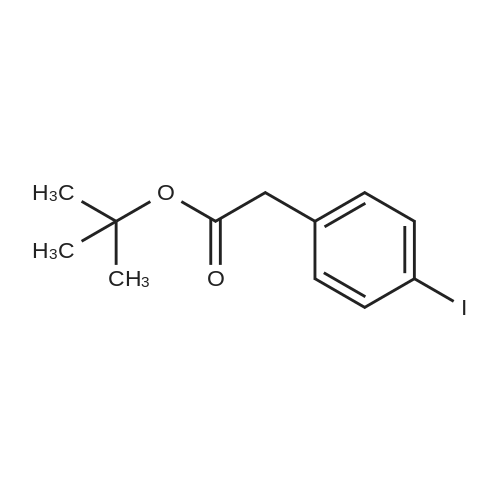
To a stirred solution of 4-iodophenylacetic acid (5.0 g, 19.1 mmol) in MeOH (200 mL) was added 4N hydrochloric acid in dioxane (10 mL). The reaction mixture was stirred for 24 h at room temperature and then the solvent was removed under reduced pressure to give the title compound (5.17 g, 98% yield), which was used without further purification. |
| 98% |
With hydrogenchloride; In 1,4-dioxane; at 20℃; for 24h; |
To a stirred solution of 4-iodophenylacetic acid (5.0 g, 19.1 mmol) in MEOH (200 mL) was added 4N hydrochloric acid in dioxane (10 mL). The reaction mixture was stirred for 24 h at room temperature and then the solvent was removed under reduced pressure to give the title compound (5.17 g, 98% yield), which was used without further purification. |
| 93% |
With thionyl chloride; at 0 - 20℃; for 1h; |
Methyl (4-iodophenyl)acetate (13).; To a solution of <strong>[1798-06-7](4-iodophenyl)acetic acid</strong> (12) (15.0 g, 57.0 mmol) in dry methanol (50 mL) was added SOCl2 (20.7 mL, 285 mmol, 5 equiv) drop wise at 00C. After stirring for 1 h at room temperature, the solvent was removed under reduced pressure and the residue dissolved in Et2O (400 mL). The organic phase was subsequently washed with saturated aqueous NaHCO3 (400 mL), saturated aqueous NH4Cl (400 mL) and brine (400 mL) and dried over MgSO4. Concentration under reduced pressure gave 13 (14.7 g, 93%). Rf=O.57 (EtOAc/hexane, 1 :4); 1H NMR (360 MHz, CDCl3) δ 7.65 (d, J=8.3 Hz, 2H), 7.03 (d, J=8.3 Hz, 2H), 3.69 (s, 3H), 3.56 (s, 3H); 13C NMR (90 MHz, CDCl3) δ 171.3, 137.6, 133.5, 131.2, 92.5, 52.0, 40.5. |
| 90% |
With tert.-butylnitrite; at 40℃; for 48h; |
Add compound 1z (0.5 mmol, 131.1 mg) and methanol containing 40 mol% tert-butyl nitrite to the reaction tube; then react for 48 hours at 40C in air; after the reaction, add sodium thiosulfate and stir. After quenching, using a rotary evaporator to remove the solvent, silica gel adsorption, and finally column chromatography with a mixed solvent of ethyl acetate and petroleum ether to obtain the product 3z, the yield is 90% |
| 86% |
With thionyl chloride; at 20℃; for 36h; |
A solution of 5 g (19 mmol) of [(4-IODO-PHENYL)-ACETIC ACID] in 50 mL of methanol [(MEOH)] was added dropwise 3.5 mL [(48] mmol) of [THIENYL] [CHLORIDE (SOCL2),] and the resulting mixture was stirred at rt for 36 h, after which time analysis by thin layer chromatography (TLC) indicated product formation. The mixture was concentrated to give 4.5 g [(86%] yield) of (4-iodo-phenyl)-acetic acid methyl ester as a pale beige oil as indicated by H NMR. A solution of 2. 88 mL (20.5 mmol) [OF DIISOPROPYLAMINE (IPR2NH)] in 50 mL of tetrahydrofuran [(THF)] was flushed with argon and cooled [TO-78 C.] To this solution was added dropwise 8.2 mL (20.5 mmol) [OF N-BUTYLLITHIUM (NBULI)] 2. 5 M solution in hexane, and the resulting mixture was stirred at-78 [C] for 20 min, after which time a solution of 4.5 g (16.3 mmol) of [(4-IODO-PHENYL)-ACETIC ACID METHYL ESTER] in 25 mL of THF was added dropwise. The mixture was allowed to warm up to rt for 40 min, then it was cooled again [TO-78 C,] and 2.62 mL (19.6 mmol) [OF CYCLOHEXANECARBONYL CHLORIDE] was added dropwise, and the resulting mixture was allowed to warm up to rt, and stirred at rt overnight under argon. The reaction mixture was quenched on ice by the addition of saturated ammonium chloride solution, and extracted with ethyl acetate. The combined organic extracts were washed with water, brine, dried over sodium sulfate and concentrated to give 7 g of a burgundy oil which was chromatographed on silica gel (Biotage; 10% ethyl acetate in hexane) to afford 5.06 g [(80%] yield) of desired [3-CYCLOHEXYL-2-(4-IODO-PHE72YL)-3-OXO-PROPIONIC ACID METHYL ESTER AS] a pale yellow solid as indicated [BY 1HNMR] (1: 2.6 keto: enol ratio). LC-MS-calcd for C16Hl9IO3 [[M++H] +] : 387.04, found: 387.0. According to a modified literature procedure (Collins, [1.] et al [J. MED. CHENA.] 2002, 45, 1887-1900) a solution of 5.06 g (13.1 mmol) of 3-cyclohexyl-2-(4-iodo-phenyl)-3-oxo- [PROPIONIC ACID METHYL ESTER] in 80 mL of dimethylsulfoxide (DMSO) was added a solution of 1.53 g (26.2 mmol) of sodium chloride (NaCI) in 5.8 mL of water (H2O), and the resulting mixture was heated at [150 C] for 3 h during which time a white solid formed. The reaction mixture was cooled to rt, poured into 500 mL of water, and extracted thoroughly with ethyl acetate. The combined organic extracts were washed with water (3 times), brine, dried over sodium sulfate and concentrated to give 4.13 g (96% yield) of desired 1-cyclohexyl-2- (4-iodo- phenyl)-ethanone as a yellow solid as indicated by 1H NMR (containing small traces of impurities). The product was used without any further purification in the next step. According to a modified literature procedure (Wasserman, H. H.; Ives, J. L. [J.] Org. Chem. 1985, 50, 3573-3580) a solution of 4.13 g (12.6 mmol) of [1-CYCLOHEXYL-2- (4-IODO-] [PHENYL)-ETHANONE] in 20 mL of toluene was flushed with argon. To this solution was added 2.7 mL (17.6 mmol) [OF METHOXY BIS (DIMETHYLAMINOQ7WLETHA7LE,] and the resulting mixture was stirred at 70C under argon overnight. The reaction mixture was concentrated to give 5.07 g of crude 1-cyclohexyl-3-dimethylamino-2-(4-iodo-phenyl)-propenone as indicated [BY'H NMR] (containing traces of starting material). The product was used without any further purification in the next step. A solution of 2.55 g (6.32 mmol max) of crude 1-cyclohexyl-3-dimethylamino-2-(4- [IODO-PHEENYL)-PROPENONE,] and 0.98 g (6.32 mmol) [OF 5-AFNINO-IH-PYRAZOLE-4-CARBOXYLIC ACID] ethyl ester in 30 mL of acetic acid [(HOAC)] was heated at reflux for 66 h, during which time a precipitate formed. The precipitate was filtered off and discarded, and the acetic acid filtrate was concentrated to a solid residue, which was washed with 1: 1 ethyl acetate: hexane and dried to give 1.67 g (55% yield) of 7-cyclohexyl-6-(4-iodo-phenyl)-pyrazolo[1,5-a]pyrimidine-3- carboxylic acid ethyl ester as a pale yellow [SOLID.'H] NMR (CDC13) [5] 8.57 (s, [1H),] 8. 53 (s, 1H), 7.86-7. 83 (d, J= 8.4 Hz, 2H), 7.1-7. 06 (d, J= 8.4 Hz, 2H), 4.46 (q, 2H, J= 7.2 Hz), 3.31- 3.21 [(M,] 1H), 2.62-2. 41 [(M,] 2H), 1.87-1. 82 [(M,] 2H), 1.74-1. 66 [(IN,] 3H), 1.44 (t, 3H, J= 7.2 Hz), 1.41-1. 21 [(M,] 3H). A solution of 1.66 g (4.1 mmol max) of crude 1-cyclohexyl-3-dimethylamino-2-(4-iodo- [PHENYL)-PROPENONE,] and 0.44 g (4.1 mmol) [OF 5-AMI7LO-LH-PYRAZOLE-4-CARBONITRILE IN] 20 mL of acetic acid [(HOAC)] was heated at reflux for [66 H,] during which time a finely dispersed precipitate formed. Since all attempts to filter off the precipitate were unsuccessful, the reaction mixture was concentrated to give a brown residue which was chromatographed on silica gel (Biotage; 2% ethyl acetate in dichloromethane) to afford 1.09 g (62% yield) of 7- cyclohexyl-6-(4-iodo-phenyl)-pyrazolo[1,5-a]pyrimidine-3-carbonitrile as a yellow solid NMR (CDC13) [6] 8.49 (s, 1H), 8.39 (s, 1H), 7.88-7. 86 (d, J= 8.4 Hz, 2H), 7.07-7. 05 (d, J= 8.4 Hz, [2H),] 4.13 (q, 2H, J= 7.2 Hz), 3.28-3. 21 [(M,] 1H), 2.59-2. 47 [(M,] 2H), 1.87-1. 8... |
|
With sulfuric acid; at 85℃; for 16h;Inert atmosphere; |
A vessel was charged with Compound Q21A (10 g, 38.1 mmol, 1.00 eq ) in MeOH (60 mL). H2SO4 (3.74 g, 38.1 mmol, 2.03 mL, 1.00 eq ) was added to the mixture under N2. The reaction was stirred at 85 C under N2 for 16 h. The reaction was concentrated under reduced pressure to give a residue. H2O (70 mL) was added, and then reaction was extracted with EtOAc (70, 50, 30 mL). The combined organic phases were washed with saturated aq NaHCCh (70, 50mL). The final organic phase was concentrated under reduced pressure to give Compound Q21B. NMR (400 MHz, CDCh) d 7.65 (d, J= 8.4 Hz, 2H), 7.04 (d, J= 8.4 Hz, 2H), 3.70 (s, 3H), 3.57 (s, 2H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping