|
With hydrogen;[Rh(S-PCyCo-BoPhoz)(COD)]OTf; In isopropyl alcohol; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 16.Table 16EntryLigandSolventConv(percent) (HPLC)Product (HPLC)d.e (percent)config1(S-Et-BoPhoz)y MeOH94832R,3S2(S-Et-BoPhoz)THFt 52512R.3S3(S-Et-BoPhoz)BOH73422R.3S4(R-Et-BoPhoz)MeOH9545L2R,3S5(R-Et-BoPhoz)DCE15792R.3S6(S-PCyCo-BoPhoz)MeOHToo632S.3S7(S-PCyCo-BoPhoz) jTHF74392R.3S8(S-PCyCo-BoPhoz)EtOH99342S.3S9(S-PCyCo-BoPhoz)'PrOH9973h 2S.3S10(S-PCyCo-BoPhoz)DCE15142R.3S11(R-PCyCo-BoPhoz)EtOH100522R.3SaReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(S-PCyCo-BoPhoz)(COD)]OTf; In ethanol; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 16.Table 16EntryLigandSolventConv(percent) (HPLC)Product (HPLC)d.e (percent)config1(S-Et-BoPhoz)y MeOH94832R,3S2(S-Et-BoPhoz)THFt 52512R.3S3(S-Et-BoPhoz)BOH73422R.3S4(R-Et-BoPhoz)MeOH9545L2R,3S5(R-Et-BoPhoz)DCE15792R.3S6(S-PCyCo-BoPhoz)MeOHToo632S.3S7(S-PCyCo-BoPhoz) jTHF74392R.3S8(S-PCyCo-BoPhoz)EtOH99342S.3S9(S-PCyCo-BoPhoz)'PrOH9973h 2S.3S10(S-PCyCo-BoPhoz)DCE15142R.3S11(R-PCyCo-BoPhoz)EtOH100522R.3SaReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(S-PCyCo-BoPhoz)(COD)]OTf; In 1,2-dichloro-ethane; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 16.Table 16EntryLigandSolventConv(percent) (HPLC)Product (HPLC)d.e (percent)config1(S-Et-BoPhoz)y MeOH94832R,3S2(S-Et-BoPhoz)THFt 52512R.3S3(S-Et-BoPhoz)BOH73422R.3S4(R-Et-BoPhoz)MeOH9545L2R,3S5(R-Et-BoPhoz)DCE15792R.3S6(S-PCyCo-BoPhoz)MeOHToo632S.3S7(S-PCyCo-BoPhoz) jTHF74392R.3S8(S-PCyCo-BoPhoz)EtOH99342S.3S9(S-PCyCo-BoPhoz)'PrOH9973h 2S.3S10(S-PCyCo-BoPhoz)DCE15142R.3S11(R-PCyCo-BoPhoz)EtOH100522R.3SaReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(S-PCyCo-BoPhoz)(COD)]OTf; In tetrahydrofuran; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 16.Table 16EntryLigandSolventConv(percent) (HPLC)Product (HPLC)d.e (percent)config1(S-Et-BoPhoz)y MeOH94832R,3S2(S-Et-BoPhoz)THFt 52512R.3S3(S-Et-BoPhoz)BOH73422R.3S4(R-Et-BoPhoz)MeOH9545L2R,3S5(R-Et-BoPhoz)DCE15792R.3S6(S-PCyCo-BoPhoz)MeOHToo632S.3S7(S-PCyCo-BoPhoz) jTHF74392R.3S8(S-PCyCo-BoPhoz)EtOH99342S.3S9(S-PCyCo-BoPhoz)'PrOH9973h 2S.3S10(S-PCyCo-BoPhoz)DCE15142R.3S11(R-PCyCo-BoPhoz)EtOH100522R.3SaReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(S-PCyCo-BoPhoz)(COD)]OTf; In methanol; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 16.Table 16EntryLigandSolventConv(percent) (HPLC)Product (HPLC)d.e (percent)config1(S-Et-BoPhoz)y MeOH94832R,3S2(S-Et-BoPhoz)THFt 52512R.3S3(S-Et-BoPhoz)BOH73422R.3S4(R-Et-BoPhoz)MeOH9545L2R,3S5(R-Et-BoPhoz)DCE15792R.3S6(S-PCyCo-BoPhoz)MeOHToo632S.3S7(S-PCyCo-BoPhoz) jTHF74392R.3S8(S-PCyCo-BoPhoz)EtOH99342S.3S9(S-PCyCo-BoPhoz)'PrOH9973h 2S.3S10(S-PCyCo-BoPhoz)DCE15142R.3S11(R-PCyCo-BoPhoz)EtOH100522R.3SaReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;(Rp)-pseudo-o-bis(di(3,5-dimethylphenyl)phosphino)[2.2]paracyclophane; bis(cycloocta-1,5-diene)rhodium(I) trifluoromethanesulfonate; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 18:Table 18EntryRh precursorConv(percent) (HPLC)Product (HPLC)d.e (percent)config1[Rh(NBD)2]BF49961^ 2R,3S2[Rh("COD)2pTf10077h2R,3S3[Rh(ethylene)2cF|29245h2R,3S4[Rh(ethyiene)2(acac)j87h 46h 2R.3S5rRh(C6j2"(OA"CJJ2431 48t 2R,3SaReaction conditions: 1mmol substrate, catalyst generated in situ from the corresponding Rh precursor and R-Xyl-PhanePhos. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;(Rp)-pseudo-o-bis(di(3,5-dimethylphenyl)phosphino)[2.2]paracyclophane; bis(ethylene)rhodium acetylacetonate; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 18:Table 18EntryRh precursorConv(percent) (HPLC)Product (HPLC)d.e (percent)config1[Rh(NBD)2]BF49961^ 2R,3S2[Rh("COD)2pTf10077h2R,3S3[Rh(ethylene)2cF|29245h2R,3S4[Rh(ethyiene)2(acac)j87h 46h 2R.3S5rRh(C6j2"(OA"CJJ2431 48t 2R,3SaReaction conditions: 1mmol substrate, catalyst generated in situ from the corresponding Rh precursor and R-Xyl-PhanePhos. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(R-Xyl-PhanePhos)(NBD)]BF4; In methanol; at 50℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
The following ligands coordinated to rhodium were chosen for initial experimental studies.OMe OMeMeO' ^"PPh2 MeO MeO^^k ^PPh2 MeOPXyl2 PXyl2Ph2P PPh2i iNHPPh2 NHPPh2H 00OMe Xyl-P-PhosH8-BINAMPSpirOPPhanephos Xyl-Phanephos MeOXyl-Phanephos Cy-Phanephos'Pr-Phanepho"'."jf ,--'""N.^MeO^^P'Pr2 FeDIPFcMe-DUPHOSMeODIPAMPSome results of this study are shown in Table 14.Table 14EntryCatalyst(°C)P(bar)Conv (percent) (HPLC)Product . (HPLC)d.econfig1[Rh(R-Xyl-PhanePhos)(NBD)]BF4501096502R.3S2[Rh(S-MeOXyl-PhanePhos)(NBD)]BF45010100562R.3S3[Rh(R-Me-DuPhos)(COD)]OTf501015682R.3S4[Rh(R-SpirOP)(NBD)]BF4501011842R.3SReaction conditions: 1mmol substrate, S/C ratio = 100/1, 4ml_ MeOH, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(S-MeOXyl-PhanePhos)(NBD)]BF4; In methanol; at 50℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
The following ligands coordinated to rhodium were chosen for initial experimental studies.OMe OMeMeO' ^"PPh2 MeO MeO^^k ^PPh2 MeOPXyl2 PXyl2Ph2P PPh2i iNHPPh2 NHPPh2H 00OMe Xyl-P-PhosH8-BINAMPSpirOPPhanephos Xyl-Phanephos MeOXyl-Phanephos Cy-Phanephos'Pr-Phanepho"'."jf ,--'""N.^MeO^^P'Pr2 FeDIPFcMe-DUPHOSMeODIPAMPSome results of this study are shown in Table 14.Table 14EntryCatalyst(°C)P(bar)Conv (percent) (HPLC)Product . (HPLC)d.econfig1[Rh(R-Xyl-PhanePhos)(NBD)]BF4501096502R.3S2[Rh(S-MeOXyl-PhanePhos)(NBD)]BF45010100562R.3S3[Rh(R-Me-DuPhos)(COD)]OTf501015682R.3S4[Rh(R-SpirOP)(NBD)]BF4501011842R.3SReaction conditions: 1mmol substrate, S/C ratio = 100/1, 4ml_ MeOH, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(R-Me-DuPhos)(COD)]OTf; In methanol; at 50℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
The following ligands coordinated to rhodium were chosen for initial experimental studies.OMe OMeMeO' ^"PPh2 MeO MeO^^k ^PPh2 MeOPXyl2 PXyl2Ph2P PPh2i iNHPPh2 NHPPh2H 00OMe Xyl-P-PhosH8-BINAMPSpirOPPhanephos Xyl-Phanephos MeOXyl-Phanephos Cy-Phanephos'Pr-Phanepho"'."jf ,--'""N.^MeO^^P'Pr2 FeDIPFcMe-DUPHOSMeODIPAMPSome results of this study are shown in Table 14.Table 14EntryCatalyst(°C)P(bar)Conv (percent) (HPLC)Product . (HPLC)d.econfig1[Rh(R-Xyl-PhanePhos)(NBD)]BF4501096502R.3S2[Rh(S-MeOXyl-PhanePhos)(NBD)]BF45010100562R.3S3[Rh(R-Me-DuPhos)(COD)]OTf501015682R.3S4[Rh(R-SpirOP)(NBD)]BF4501011842R.3SReaction conditions: 1mmol substrate, S/C ratio = 100/1, 4ml_ MeOH, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(S-Et-BoPhoz)(COD)]OTf; In ethanol; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 16.Table 16EntryLigandSolventConv(percent) (HPLC)Product (HPLC)d.e (percent)config1(S-Et-BoPhoz)y MeOH94832R,3S2(S-Et-BoPhoz)THFt 52512R.3S3(S-Et-BoPhoz)BOH73422R.3S4(R-Et-BoPhoz)MeOH9545L2R,3S5(R-Et-BoPhoz)DCE15792R.3S6(S-PCyCo-BoPhoz)MeOHToo632S.3S7(S-PCyCo-BoPhoz) jTHF74392R.3S8(S-PCyCo-BoPhoz)EtOH99342S.3S9(S-PCyCo-BoPhoz)'PrOH9973h 2S.3S10(S-PCyCo-BoPhoz)DCE15142R.3S11(R-PCyCo-BoPhoz)EtOH100522R.3SaReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(S-Et-BoPhoz)(COD)]OTf; In tetrahydrofuran; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 16.Table 16EntryLigandSolventConv(percent) (HPLC)Product (HPLC)d.e (percent)config1(S-Et-BoPhoz)y MeOH94832R,3S2(S-Et-BoPhoz)THFt 52512R.3S3(S-Et-BoPhoz)BOH73422R.3S4(R-Et-BoPhoz)MeOH9545L2R,3S5(R-Et-BoPhoz)DCE15792R.3S6(S-PCyCo-BoPhoz)MeOHToo632S.3S7(S-PCyCo-BoPhoz) jTHF74392R.3S8(S-PCyCo-BoPhoz)EtOH99342S.3S9(S-PCyCo-BoPhoz)'PrOH9973h 2S.3S10(S-PCyCo-BoPhoz)DCE15142R.3S11(R-PCyCo-BoPhoz)EtOH100522R.3SaReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(R-Xyl-PhanePhos)(COD)]OTf; In butan-1-ol; at 50℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 17.Table 17EntryLigandSolventConv (percent) (HPLC)Product (HPLC)d.e (percent)config1(R-Xyl-PhanePhos)MeOH100632R,3S2(R-Xyl-PhanePhos)EtOH100772R,3S3(R-Xyl-PhanePhos)10percentH2O-EtOH100802R,3S4(R-Xyl-PhanePhos)1-BuOH100792R,3S5(R-Xyl-PhanePhos)10percent H20-BuOH100842R.3SReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4mL solvent, 50°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(R-Xyl-PhanePhos)(COD)]OTf; In ethanol; at 50℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 17.Table 17EntryLigandSolventConv (percent) (HPLC)Product (HPLC)d.e (percent)config1(R-Xyl-PhanePhos)MeOH100632R,3S2(R-Xyl-PhanePhos)EtOH100772R,3S3(R-Xyl-PhanePhos)10percentH2O-EtOH100802R,3S4(R-Xyl-PhanePhos)1-BuOH100792R,3S5(R-Xyl-PhanePhos)10percent H20-BuOH100842R.3SReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4mL solvent, 50°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(R-Xyl-PhanePhos)(COD)]OTf; In methanol; at 50 - 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
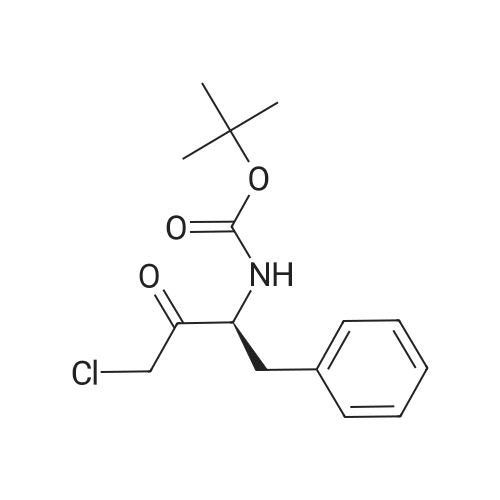
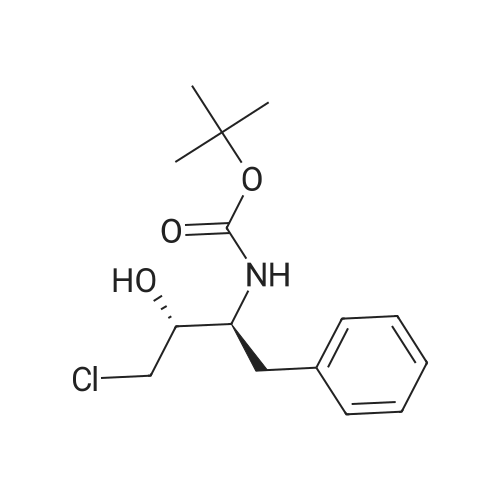
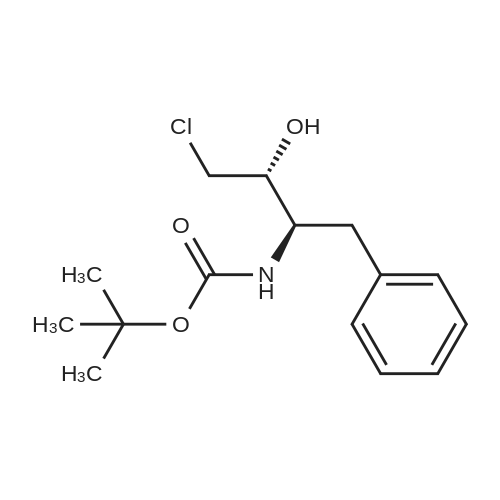
The following ligands coordinated to rhodium were chosen for experimental studies:PV ,Ph PPPh2Phv fh P,PPh2 -EtPh PPh PCy2Ph. fhPPh,OY~Me ^MN~Et 0-AEt ^AHFeFeFeFeMeBoPhozEtBoPhozPCycoBoPhozProBoPhozSome results of this study are shown in Table 15.Table 15EntryLigandSolventConv(percent) (HPLC)Product (HPLC).d:e (percent) J .cqnfjg1lS-Me:BoPhc)z)MeOH98722R,3S(S-Et-BoPhoz)MeOH94832R,3SIR-Xy^PhjmePhos^MeOH10059 2R.3SReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4mL MeOH, 65°C, 10 bar, unoptimized reaction time 20 hrs.; Example 17Influence of the solvent on the hvdrogenation of BocChloroketone in the presence of fRh(COD21QTf/Xvl-PhanePhos systemsSome results of this study are shown in Table 17.Table 17EntryLigandSolventConv (percent) (HPLC)Product (HPLC)d.e (percent)config1(R-Xyl-PhanePhos)MeOH100632R,3S2(R-Xyl-PhanePhos)EtOH100772R,3S3(R-Xyl-PhanePhos)10percentH2O-EtOH100802R,3S4(R-Xyl-PhanePhos)1-BuOH100792R,3S5(R-Xyl-PhanePhos)10percent H20-BuOH100842R.3SReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4mL solvent, 50°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(R-Et-BoPhoz)(COD)]OTf; In 1,2-dichloro-ethane; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 16.Table 16EntryLigandSolventConv(percent) (HPLC)Product (HPLC)d.e (percent)config1(S-Et-BoPhoz)y MeOH94832R,3S2(S-Et-BoPhoz)THFt 52512R.3S3(S-Et-BoPhoz)BOH73422R.3S4(R-Et-BoPhoz)MeOH9545L2R,3S5(R-Et-BoPhoz)DCE15792R.3S6(S-PCyCo-BoPhoz)MeOHToo632S.3S7(S-PCyCo-BoPhoz) jTHF74392R.3S8(S-PCyCo-BoPhoz)EtOH99342S.3S9(S-PCyCo-BoPhoz)'PrOH9973h 2S.3S10(S-PCyCo-BoPhoz)DCE15142R.3S11(R-PCyCo-BoPhoz)EtOH100522R.3SaReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(R-Et-BoPhoz)(COD)]OTf; In methanol; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 16.Table 16EntryLigandSolventConv(percent) (HPLC)Product (HPLC)d.e (percent)config1(S-Et-BoPhoz)y MeOH94832R,3S2(S-Et-BoPhoz)THFt 52512R.3S3(S-Et-BoPhoz)BOH73422R.3S4(R-Et-BoPhoz)MeOH9545L2R,3S5(R-Et-BoPhoz)DCE15792R.3S6(S-PCyCo-BoPhoz)MeOHToo632S.3S7(S-PCyCo-BoPhoz) jTHF74392R.3S8(S-PCyCo-BoPhoz)EtOH99342S.3S9(S-PCyCo-BoPhoz)'PrOH9973h 2S.3S10(S-PCyCo-BoPhoz)DCE15142R.3S11(R-PCyCo-BoPhoz)EtOH100522R.3SaReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;(Rp)-pseudo-o-bis(di(3,5-dimethylphenyl)phosphino)[2.2]paracyclophane; diacetatetetracarbonyl dirhodium(I); at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 18:Table 18EntryRh precursorConv(percent) (HPLC)Product (HPLC)d.e (percent)config1[Rh(NBD)2]BF49961^ 2R,3S2[Rh("COD)2pTf10077h2R,3S3[Rh(ethylene)2cF|29245h2R,3S4[Rh(ethyiene)2(acac)j87h 46h 2R.3S5rRh(C6j2"(OA"CJJ2431 48t 2R,3SaReaction conditions: 1mmol substrate, catalyst generated in situ from the corresponding Rh precursor and R-Xyl-PhanePhos. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;(Rp)-pseudo-o-bis(di(3,5-dimethylphenyl)phosphino)[2.2]paracyclophane; bis(ethylene)rhodium(I) chloride dimer; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 18:Table 18EntryRh precursorConv(percent) (HPLC)Product (HPLC)d.e (percent)config1[Rh(NBD)2]BF49961^ 2R,3S2[Rh("COD)2pTf10077h2R,3S3[Rh(ethylene)2cF|29245h2R,3S4[Rh(ethyiene)2(acac)j87h 46h 2R.3S5rRh(C6j2"(OA"CJJ2431 48t 2R,3SaReaction conditions: 1mmol substrate, catalyst generated in situ from the corresponding Rh precursor and R-Xyl-PhanePhos. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;(Rp)-pseudo-o-bis(di(3,5-dimethylphenyl)phosphino)[2.2]paracyclophane; rhodium(I)-bis(1,5-cyclooctadiene) tetrafluoroborate; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
Some results of this study are shown in Table 18:Table 18EntryRh precursorConv(percent) (HPLC)Product (HPLC)d.e (percent)config1[Rh(NBD)2]BF49961^ 2R,3S2[Rh("COD)2pTf10077h2R,3S3[Rh(ethylene)2cF|29245h2R,3S4[Rh(ethyiene)2(acac)j87h 46h 2R.3S5rRh(C6j2"(OA"CJJ2431 48t 2R,3SaReaction conditions: 1mmol substrate, catalyst generated in situ from the corresponding Rh precursor and R-Xyl-PhanePhos. S/C ratio = 100/1, 4ml_ solvent, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With hydrogen;[Rh(S-Me-BoPhoz)(COD)]OTf; In methanol; at 65℃; under 7500.75 Torr; for 20h;Argonaut Endeavour hydrogenation unit;Product distribution / selectivity; |
The following ligands coordinated to rhodium were chosen for experimental studies:PV ,Ph PPPh2Phv fh P,PPh2 -EtPh PPh PCy2Ph. fhPPh,OY~Me ^MN~Et 0-AEt ^AHFeFeFeFeMeBoPhozEtBoPhozPCycoBoPhozProBoPhozSome results of this study are shown in Table 15.Table 15EntryLigandSolventConv(percent) (HPLC)Product (HPLC).d:e (percent) J .cqnfjg1lS-Me:BoPhc)z)MeOH98722R,3S(S-Et-BoPhoz)MeOH94832R,3SIR-Xy^PhjmePhos^MeOH10059 2R.3SReaction conditions: 1mmol substrate, [Rh(bisphosphine)(COD)]OTf generated in the corresponding solvent by reacting [Rh(COD)2]OTf with the bisphosphine for 30min under N2. S/C ratio = 100/1, 4mL MeOH, 65°C, 10 bar, unoptimized reaction time 20 hrs. |
|
With isopropyl alcohol;aluminum tri-tert-butoxide; In ethyl acetate;Inert atmosphere;Product distribution / selectivity; |
The MPV reduction was conducted in various organic solvents as described in Example 3, where the solvent of interest was used in the place of toluene. Isopropanol (10percent) was added to each solvent for the reaction to proceed in a reasonable time.Table 1 shows the results of these reactions. The (R,S)/(S,S) ratio increased when the reaction was run in aprotic polar solvents like ethyl acetate and THF. While not limiting any embodiment by theory, it is believed that the hydrogen bonding between ketone 1 and Al(OiPr)3 may contribute to increasing the rate of reaction by keeping 1 coordinated with the aluminum center, bringing to close proximity the two reactive centers.Table 1. MPV reduction of 1 in various organic solvents |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping