| 99% |
|
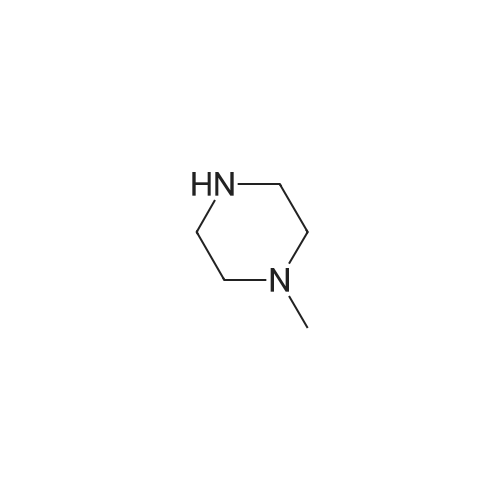
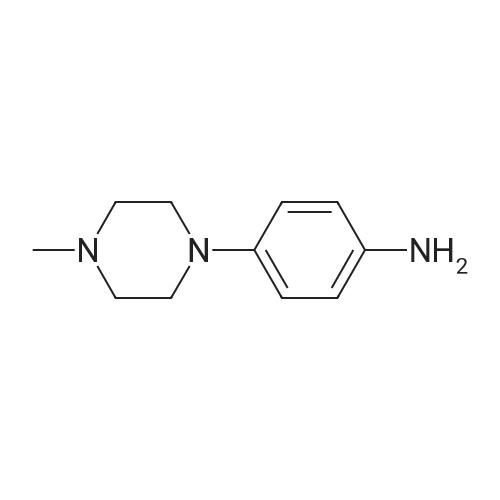
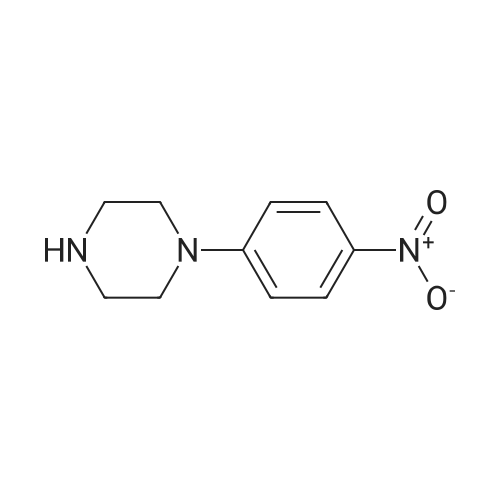
General procedure: To a solution of 7a (300 mg, 1.35 mmol), Pd-C (30 mg) in ethanol (20 mL) was added, and the mixture was stirred under reflux for 1 h. Then, hydrazine (2.6 mL) was added dropwise to the mixture and stirred over reflux for an additional 2 h. After the completion of the reaction, the reaction mixture was filtered in Celite and concentrated to give 4a. 4.2.12. 4-(4-Methylpiperazin-1-yl)aniline (4a) Black solid, yield 99%, mp 128-130 C. 1H NMR (400 MHz, CDCl3) δ 6.80 (t, J=9.1 Hz, 1H), 6.48-6.33 (m, 2H), 3.53 (s, 2H), 3.10-2.83 (m, 4H), 2.66-2.54 (m, 4H), 2.34 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 144.55, 140.23, 118.67, 116.29, 77.16, 55.39, 50.89, 46.17. ESI/MS for (C11H17N3 [M+H]+). Calcd: 192.2. Found: 191.9. |
| 99% |
With palladium 10% on activated carbon; hydrazine hydrate monohydrate; In ethanol; at 78 - 80℃; for 0.5h;Inert atmosphere; |
General procedure: The mixture of 16 (10.0 mmol) and Pd/C (10 wt%, 0.2 g) in EtOH(200 mL) was heated to reflux under argon. Then hydrazine hydrate(85 wt%, 200.0 mmol) was added dropwise to the mixture over 0.5 hmaintaining the temperature at 78-80 C. After the addition wascompleted, the reaction mixture was stirred and refluxed for 2 h. Thehot solution was then filtered by vacuum through celite pad. The solventwas concentrated to provide the target compounds without furtherpurificationas. |
| 98% |
With hydrogen;palladium 10% on activated carbon; In ethyl acetate; at 20℃; for 18h;Inert atmosphere; |
1-Methyl-4-(4-nitrophenyl)piperazine (111) (0.632 g, 2.86 mmol) was dissolved in EtOAc (45 mL) under an atmosphere of nitrogen and a slurry of 10% Pd/C (0.200 g) in EtOAc (5 mL) was added. The resulting suspension was then stirred vigorously under an atmosphere of hydrogen at room temperature for 8 hours. The cataiyst was removed by filtration through Celite, which was washed with EtOAc (7x 0 mL) and the solvent was removed in vacuo to give the title compound (112) (0.537 g, 98% yield) as a pink solid; H NMR (400 MHz, de-DMSO) δ 6.70 - 6.64 (m, 2H), 6.51 - 6.45 (m, 2H), 4.54 (s, 2H), 2.94 - 2.84 (m, 4H), 2.46 - 2.36 (m, 4H), 2.19 (s, 3H). LCMS Method C: rt 0.98 min; m/z 192.3 [M+H]+. |
| 98% |
With palladium 10% on activated carbon; hydrogen; In ethyl acetate; at 20℃; for 18h;Inert atmosphere; |
1-Methyi-4-(4-nitrophenyi)piperazine (111) (0.632 g, 2.86 mmol) was dissolved in EtOAc (45 mL) under an atmosphere of nitrogen and a slurry of 10% Pd/C (0.200 g) in EtOAc (5 mL) was added. The resulting suspension was then stirred vigorously under an atmosphere of hydrogen at room temperature for 18 hours. The catalyst was removed by filtration through Ceiite, which was washed with EtOAc (7x10 mL) and the solvent was removed in vacuo to give the title compound (112) (0.537 g, 98% yield) as a pink solid; 1H NMR (400 MHz, oV-DMSO) δ 6.70 - 6.64 (m, 2H), 6.51 - 6.45 (m, 2H), 4.54 (s, 2H), 2,94 - 2.84 (m, 4H), 2.46 - 2.36 (m, 4H), 2.19 (s, 3H). LCMS Method C: rt 0.98 min; m/z 192.3 [M+H]+. |
| 97% |
With iron(0); ammonia hydrochloride; In ethanol; for 5h;Reflux; |
General procedure: To a stirring solution of compound 4a (1.2g, 4.77mmol) in 70% ethanol (20mL) were added ammonium chloride (1.28g, 23.85mmol) and reduced iron powder (0.8g, 14.31mmol). The reaction was refluxed for 5h. Upon completion, the mixture was filtered while hot and rinsed with ethyl acetate. The solvent was removed in vacuo, and the residue was purified by silica gel column chromatography (PE : EA=1 : 2) to afford compound 5a as an orange solid. Yield: 97%; mp: 108-110C; 1H NMR (300MHz, DMSO-d6) δ 6.67 (d, J=8.8Hz, 2H), 6.48 (d, J=8.8Hz, 2H), 4.53 (s, 2H), 2.92-2.84 (m, 4H), 2.46-2.36 (m, 4H), 2.19 (s, 3H). MS (m/z): [M+H]+ 192.3. |
| 97% |
With palladium 10% on activated carbon; hydrogen; In methanol; ethyl acetate; for 24h; |
To a stirred solution of 10-methyl-4’-(4-nitrophenyl)piperazine (5.75 g, 26.0 mmol) in 1:1MeOH:EtOAc (60 mL) was added 10% palladium on carbon (0.058 g,10% w/w) slowly. The mixture was then stirred under an atmosphereof hydrogen for 24 h before being filtered through Celite andwashed with further MeOH. The solvent was removed in vacuo togive the title compound 19 (4.84 g, 97%) as a maroon solid. m.p.85-87 C. δH (400 MHz, CDCl3) 2.33 (3H, s, CH3), 2.57 (4H, t,J 4.8 Hz, H-30 , H-50), 3.06 (4H, t, J 4.8 Hz, H-20, H-60), 3.41 (2H, brs, NH2), 6.64 (2H, d, J 8.6 Hz, H-2, H-6), 6.81 (2H, d, J 8.6 Hz, H-3,H-5). The spectroscopic values were in agreement with literature[27]. |
| 95% |
With palladium 10% on activated carbon; hydrogen; In ethanol; at 20℃; for 9h;Inert atmosphere; Schlenk technique; |
General procedure: The nitrophenyl analogue 7a-7e (7.5 mmol) was dissolved in ethanol (50 mL), and to this solution was added 10% Pd/C (0.2 g). The reaction mixture was stirred at room temperature under an atmosphere of H2 for 9 h. After completion of the reaction, the resulting mixture was filtered through Celite. The filtrate was concentrated under high vacuum to afford the desired aniline derivatives 8a-8e. |
| 93% |
With ammonia hydrochloride; zinc powder; In tetrahydrofuran; methanol; at 27℃; for 2h; |
<strong>[16155-03-6]1-methyl-4-(4-nitrophenyl)piperazine</strong> (46, 5.1 g, 22.0 mmol) was taken in a 500 mL round bottom flask in a mixture of solvent MeOH and THF (100 mL, 1:1). NH4Cl (12.0 g, 220.0 mmol) and Zn dust (14.7 g, 220.0 mmol) were added and the reaction mixture was stirred at room temperature for 2 h. It was then filtered through sintered funnel with a pad of celite, washed with MeOH (50 mL) and concentrated under reduced pressure. The crude was further washed with n-pentane to afford 47 as a gummy residue (4.0 g, 93% yield).1H NMR (400 MHz, DMSO-d6): δ 7.34 (bs, 2H), 6.75-6.73 (m, 2H), 6.56-6.54 (m, 2H), 3.20 (m, 8H), 2.73 (s, 3H). |
| 93% |
With palladium 10% on activated carbon; hydrogen; In ethyl acetate; at 20℃; for 14h; |
12.5 g (56.5 mmol) of the compound prepared in step 5) was diluted with 120 ml of ethyl acetate, and thereafter 1.2 g (10wt %) of palladium/carbon was added thereto at room temperature and stirred under hydrogen gas for 14 hours. After the reaction was completed, the reaction mixture was washed with ethyl acetate and filtered through a Celite-filled filter under reduced pressure, and the resulting filtrate was distilled under reduced pressure to obtain 10 g of the compound of the title (Yield: 93%). 1H-NMR (300MHz, DMSO-d6) δ 6.65 (d, 2H), 6.47 (d, 2H), 4.55 (brs, 2H), 2.87 (m, 4H), 2.40 (m, 4H), 2.18 (s, 3H). |
| 92% |
With hydrogen;palladium on activated charcoal; In methanol; at 20℃; under 760.051 Torr; |
Step b - 4-(4-methylpiperazin-1-yl)benzenamine; A solution of <strong>[16155-03-6]1-methyl-4-(4-nitrophenyl)piperazine</strong> (1.03g, 4.66mmol) in MeOH (100ml) was hydrogenated at 2O0C at atmospheric pressure using an H-Cube (flow rate at 1ml/min and full hydrogen mode) using a Pd/C cartridge. The solvent was removed in vacuo to afford 4-(4-methylpiperazin-1-yl)benzenamine (0.82g, 4.29mmol, 92%) as an off-white solid. 1H NMR (CDCI3) δ 2.37 (3H, s), 2.62 (4H, t), 3.09 (4H, t), 3.40 (2H, br. s), 6.65 (2H, d), 6.82 (2H, d). LCMS (1) Rt: 0.98min; m/z (ES+) 192. |
| 91.1% |
With iron(0); ammonia hydrochloride; In ethanol; for 5h;Reflux; |
In 50ml single-necked flask M-30.60g (0.27mmol), ammonium chloride 0.46g (1.34mmol) and 70% ethanol 25ml, was added portionwise stir reduced iron powder 0.46g (0.80mmol), was heated to reflux. 5After h TLC showed the starting material is no longer remaining. Hot filtration, the filtrate was evaporated under reduced pressure, water was added, extracted with ethyl acetate (15ml × 3), the combined organic phase was washed twice with saturated brine, dried over anhydrous MgSO 4. After concentration under reduced pressure column chromatography (PE:EA = 1:1) was a light brown liquid 0.47g, yield 91.1%. |
| 89% |
With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; |
To a 500 mL reaction flask,Adding 13.12 g of compound 20,Add 200 mL of methanol,And 1.45 g of 10% Pd / C,Through the hydrogen, room temperature reaction overnight,The reaction ends,The palladium-carbon was removed by filtration,The filtrate was concentrated,The residue was purified by column chromatography (MeOH: DCM = 10: 1)To give 9.9 g of a gray solid 7-2. Yield 89%. |
| 86.8% |
With palladium 10% on activated carbon; hydrogen; In methanol; lithium hydroxide monohydrate; ethyl acetate; at 20℃; for 2h; |
To a stirred solution of 1 -methyl-4-(4-nitrophenyl)piperazine (3) (1 g, 4.519 mmol, 1 eq) in mixture of MeOH: EtOAc: 0 (5: 4: 2 mL) was added 10% Pd/C (300 mg) under inert atmosphere at room temperature into a hydrogenator. The mixture was degassed for 15 min with the help of alternative vacuum and nitrogen. The reaction was stirred under hydrogen atmosphere for 2 h at room temperature. The reaction was monitored by TLC (M.Ph: 5% methanol in DCM). The reaction mixture was filtered through C6lite bed and the filtrate was concentrated in vacuo to afford 4 (750 mg, 86.8%) as yellow solid. NMR (DMSO-*, 400 MHz): δ ppm 6.75 (d, .7=8.80 Hz, 2H), 6.56 (d, 7=8.80 Hz, 2H), 4.63 (hr. s, 2H), 2.93-3.02 (m, 4H), 2.46-2.54 (m, 4H), 2.28 (s, 3H); LC-MS: m/z 191.80 [M+H]+. |
| 86.8% |
With palladium 10% on activated carbon; hydrogen; In methanol; lithium hydroxide monohydrate; ethyl acetate; at 20℃; for 2h; |
To a stirred solution of 1 -methyl-4-(4-nitrophenyl)piperazine (3) (1 g, 4.519 mmol, 1 eq) in mixture of MeOH: EtOAc: 0 (5: 4: 2 mL) was added 10% Pd/C (300 mg) under inert atmosphere at room temperature into a hydrogenator. The mixture was degassed for 15 min with the help of alternative vacuum and nitrogen. The reaction was stirred under hydrogen atmosphere for 2 h at room temperature. The reaction was monitored by TLC (M.Ph: 5% methanol in DCM). The reaction mixture was filtered through C6lite bed and the filtrate was concentrated in vacuo to afford 4 (750 mg, 86.8%) as yellow solid. NMR (DMSO-*, 400 MHz): δ ppm 6.75 (d, .7=8.80 Hz, 2H), 6.56 (d, 7=8.80 Hz, 2H), 4.63 (hr. s, 2H), 2.93-3.02 (m, 4H), 2.46-2.54 (m, 4H), 2.28 (s, 3H); LC-MS: m/z 191.80 [M+H]+. |
| 83.2% |
With palladium 10% on activated carbon; In methanol; at 20℃; for 5h; |
1.0 g of 4- (4-methylpiperazine) nitrobenzene (compound 6) was dissolved in 20 mL of methanol, Slowly added with stirring catalytic amount of 10% Pd / C, at room temperature for 5h. The reaction solution was suction filtered to remove Pd / C,Wash the filter cake with methanol. The filtrate was depressurized to remove methanol and extracted with ethyl acetate (30 mL × 3). The organic phases were combined,Saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure,0.72 g of 4- (4-methylpiperazine) aniline (Compound 7) was isolated by column chromatography, and the reaction yield was 83.2%. |
| 83.7% |
With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; for 5h; |
1.0 g of 4- (4-methylpiperazine) nitrophenylhydrazone was dissolved in 10 mL of methanol, and a catalytic amount of 10% Pd / C (mass fraction) was slowly added under stirring, and hydrogen was bubbled in, and reacted at room temperature for 5 h. The reaction solution was suction filtered and the filter cake was washed with methanol. The filtrate was spin-dried under reduced pressure to remove methanol, and the residue was separated by silica gel column chromatography to obtain 0.72 g of 4- (4-methylpiperazine) aniline (7) as an off-white solid with a yield of 83.7%. |
| 83.7% |
With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; for 5h; |
Dissolve 1.0 g of <strong>[16155-03-6]4-(4-methylpiperazine)nitrobenzene</strong> (6) in 10 mL of methanol, slowly add a catalytic amount of 10% Pd / C (mass fraction) under stirring and pass hydrogen, and react at room temperature for 5 h. the reaction solution was suction filtered and the filter cake was washed with methanol. The filtrate was spin-dried under reduced pressure to remove methanol, and the residue was separated by silica gel column chromatography to obtain 0.72 g of 4-(4-methylpiperazine)aniline (7) as an off-white solid, with a yield of 83.7%. |
| 80% |
With iron(0); ammonia hydrochloride; In ethanol; for 8h;Reflux; |
Add in 100ml single neck bottleM-3 (2.00 g, 9.04 mmol),Ammonium chloride (2.42g, 45.2mmol)And 70% ethanol 25ml,Stir well and add in batchesReduced iron powder (1.52g, 27.1mmol),Heat to reflux.After 8 h, TLC showed no residue of starting material.Hot and suction filtration,The filtrate was distilled off under reduced pressure.Crude column chromatography (PE: EA = 1: 1)A pale yellow liquid of 1.38 g was obtained in a yield of 80.0%. |
| 78.4% |
With ammonia hydrochloride; zinc powder; In tetrahydrofuran; methanol; at 0 - 20℃; for 18h; |
General procedure: Nitro compound 2a-r (3.0 mmol, 1 eq) was dissolved in amixture of THF (9 mL) and MeOH (6 mL) at 0 C. Subsequently, zincpowder (15.0 mmol, 5 eq), followed by ammonium chloride(15.0 mmol, 5 eq) were added and the reaction mixture was stirredat room temperature for 18 h. After completion of the reaction, theresulting mixture was filtered through Celite and the filtrate wasconcentrated in vacuo. The crude was purified by flash chromatographywith 97:3 (v/v) dichloromethane - methanol. |
| 75% |
With ammonium hydroxide; sodium dihydrosulfite; In lithium hydroxide monohydrate; for 0.25h;Reflux; |
General procedure: To a solution of 1-(substituted) 4-nitrobenzene IVa,b,e,f(0.01 mol) in NH4OH (20 mL, 30%), a solution of sodium dithionite(7 g, 0.04 mol) in water (30 mL) was quickly added, the reactionmixture was refluxed for 15 min. After cooling, the crude productwas filtered, washed and crystallized from methylene chloride toyield target compounds Va,b,e,f. 4-(4-Methylpiperazin-1-yl) aniline Ve Yield 75% as off white acicular solid, mp 90 C, (as reported) [62,67,68]. |
| 75.6% |
With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; for 16h; |
To a stirred solution of <strong>[16155-03-6]1-methyl-4-(4-nitrophenyl)piperazine</strong> 26 (10.0 g, 45.19 mmol) in methanol (100 mL) was added 10% Pd/C (3.00 g) at room temperature under argon atmosphere. The reaction mixture was further stirred under 112 atmosphere at room temperature for 16 h. After completion of the reaction (monitored by TLC), the reaction mixture was filtered through a pad of Celite and the filtrate was concentrated under reduced pressure to afford 6.50 g (75.6% yield) of 27 as grey solid. 1H NMR (400 MHz, DMSO-d6) δ: 6.81 (d, J=8.31 Hz, 2H), 6.62 (d, J=8.31 Hz, 2H), 4.69 (br, s, 2H), 2.99-3.07 (m, 4H), 2.64 (br, s, 2H), 2.52-2.59 (m, 4H), 2.34 (s, 3H). |
| 70% |
With hydrogenchloride; tin; In chloroform; lithium hydroxide monohydrate;Reflux; |
General procedure: Concisely,1-(4-Nitro phenyl)-4-substitued piperazine derivatives (1-18) were refluxed for 3-4 h in chloroform (20 ml) with theSn/HCl solution. The solution was prepared before by dissolving tin (15mM) in30ml Con. HCl. The reaction mixture was cooled under tap water and neutralizedwith 15% NaOH solution. The resulting compound was extracted with ethylacetate. The organic layer was dried over anhydrous sodium sulphate andevaporated under reduced pressure to afford of 4-(4-substitutedpiperazin-1-yl)-phenylamine derivatives (19-36)as solid compounds. |
| 64% |
With hydrogen;palladium 10% on activated carbon; In methanol; |
l-Methyl-4-(4-nitro-phenyl)-piperazine (200mg) was hydro genated over 10% Pd-C (20mg) in methanol (20ml) at atmospheric pressure until no further gas uptake was observed. The reaction mixture was then filtered over celite and concentrated to give a crude solid. Column chromatography over silica gel using 4% methanol in dichloromethane gave 4-(4-methyl-piperazin-l-yl)-phenylamine (HOmg, 64%) as solid. |
|
palladium monocarbide; In methanol; ethanol; |
Step B-Preparation of 4-methyl-1-(4-aminophenyl)piperazine 4-Methyl-1-(4-nitrophenyl)piperazine (2.0 g, 9 mmol, Step A) and 10% Pd/C (200 mg) were added to EtOH/MeOH (1:1) (50 ml) at RT. The reaction stirred under a H2 atmosphere (via balloon) overnight. The mixture was filtered through a plug of Celiteo and the filtrate was concentrated under reduced pressure to leave the desired material as a light yellow oil. The material was used in subsequent reaction without purification. MS: (ES+) 192 (M+1)+; (ES-): 190 (M-1)-. Calc'd for C11H17N3: 191.14. |
|
palladium monocarbide; In methanol; ethanol; |
Step B-Preparation of 4-methyl-1-(4-aminophenyl)piperazine 4-Methyl-1-(4-nitrophenyl)piperazine (2.0 g, 9 mmol, Step A) and 10% Pd/C (200 mg) were added to EtOH/MeOH (1:1) (50 ml) at RT. The reaction stirred under a H2 atmosphere (via balloon) overnight. The mixture was filtered through a plug of Celite and the filtrate was concentrated under reduced pressure to leave the desired material as a light yellow oil. The material was used in subsequent reaction without purification. MS: (ES+) 192 (M+1)+; (ES-): 190 (M-1)-. Calc'd for C11H17N3: 191.14. |
|
With hydrogen;palladium 10% on activated carbon; In methanol; ethanol; at 20℃; |
Step B-Preparation of 4-methyl-1-(4-aminophenyl)piperazine 4-Methyl-1-(4-nitrophenyl)piperazine (2.0 g, 9 mmol, Step A) and 10% Pd/C (200 mg) were added to EtOH/MeOH (1:1) (50 ml) at RT. The reaction stirred under a H2 atmosphere (via balloon) overnight.The mixture was filtered through a plug of Celite and the filtrate was concentrated under reduced pressure to leave the desired material as a light yellow oil.The material was used in subsequent reaction without purification. MS: (ES+) 192 (M+1)+; (ES-): 190 (M-1)-. Calc'd for C11H17N3: 191.14. |
|
With hydrogen;palladium 10% on activated carbon; In methanol; at 20℃; for 16h; |
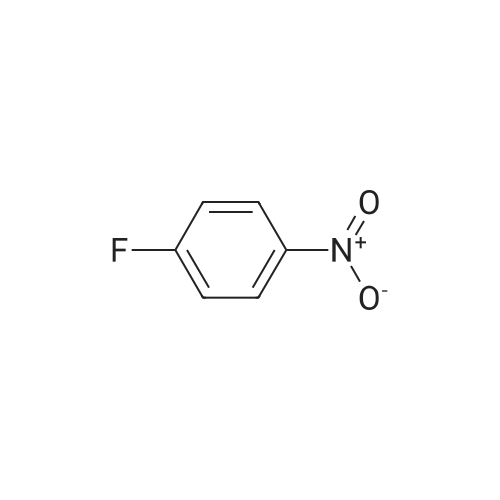
F. 4-(4-Methyl-piperazin-1-yl)-phenylamine Potassium carbonate (1.9 g, 14.2 mmol) was added to a mixture of 1-fluoro-4-nitrobenzene (1 g, 7.1 mmol) and 1-methyl-piperazine (0.94 mL, 8.5 mmol) in methyl sulfoxide (DMSO, 5 mL). The mixture was stirred at 80 C. for 3 hours. After cooling down, the residue was extracted into EtOAc. The organic layer was washed with water, brine and then dried with Na2SO4. Removal of the solvent in vacuo yielded an orange solid. The solid was dissolved in 25 mL of methanol and palladium on carbon (10% Pd/C, 50 mg) was added slowly. The system was sealed and blanketed with hydrogen. The mixture was stirred at rt for 16 hours under hydrogen. The catalyst was filtered through a celite pad and the solvent was evaporated to leave a dark purple solid (1.3 g, 80%). 1H NMR (300 MHz, CD3OD) δ (ppm): 6.90 (m, 2H), 6.81 (m, 2H), 3.38 (m, 4H), 3.26 (m, 4H), 2.93 (s, 3H). |
|
With hydrogen;nickel; In methanol; |
Step B: 44.3 g (0.2 Mol) of [1-METHYL-4- (4-NITRO-PHENYL)-PIPERAZINE] is dissolved in 1200 mL of CH30H and subjected to catalytic hydration at rt using Raney-Ni (10 g) as catalyst. After filtration over Celite, the crude product is purified via solid-distillation (0.16 mbar, [180C] ; heat temp [125C)] to obtain [4- (4-METHYL-PIPERAZIN-1-YI)-PHENYLAMINE. TITLE] compound: ES-MS: 192.1 [M+H] + ; single peak at tR= 1.08 min (System 1); Rf = 0.33 (CH2CI2/MeOH 4: 1). |
| 7.67 g |
With 10% palladium on activated carbon; hydrogen; In ethanol; at 20℃; |
The mixture of 1-methyl-4-(4-nitro-phenyl)-piperazine (10.2 g, 45.0 mmol) and 10% Pd/C (0.5g) in EtOH (350 mL) was stirred at room temperature under H2. After the completion of the reaction, the mixture was filtered through celite and purified with column chromatography (dichloromethane : MeOH : n-hexane = 10 : 1 : 1) to afford 4-(N-methyl-piperazin-1-yl)-phenylamine 9 (7.67 g, 39.8 mmol): 1H NMR (200 MHz, CDCl3) δ 2.34 (s, 3H, NCH3), 2.55 - 2.60 (m, 4H, NCH2 x 2), 3.04 - 3.09 (mz, 4H, NCH2 x 2), 3.41 (br s, 2H, NH2), 6.64 (m, 2H, ArH), 6.80 (m, 2H, ArH); MS(EI) m/e 191 [M+]. |
|
With palladium 10% on activated carbon; hydrogen; under 2585.81 Torr; |
General procedure: The substituted nitrobenzenes were reduced to the corresponding anilines by catalytic reduction (50 psi, 10% Pd/C (5% w/w)) on a Parr hydrogenator as previously described [19] The anilines (not characterized) were then condensed with 6,9-dichloro-2-methoxyacridine or 4,7-dichloroquinoline (Section 6.2.) to give 1-3, 10. |
|
With hydrogen;palladium on activated charcoal; In methanol; for 24h; |
(4-Aminophenyl)[2-(dimethylamino)ethyl]ethylamine 1-Methyl-4-(4-nitrophenyl)piperazine (7.7 g, 34.8 mmol) was dissolved in MeOH (200 mL) and Pd/C (0.185 g, 1.740 mmol) was added. The reaction mixture was placed under an atmosphere of H2 for 24 hour. The reaction was filtered through a pad of celite, washed with MeOH, concentrated and dried on high vacuum to give the product as a brown solid 6.6g. |
|
With hydrazine hydrate monohydrate; In ethanol; at 50℃; |
General procedure: o a mixture of amine 38 and N2H4-H2O (5 eq) in EtOH was added Ni Ra (0.07 eq). The suspension was heated at 50 C overnight, filtered via celite, the celite was additionally washed with EtOH. The filtrate was evaporated and dried under vacuum to provide piperazines 34a-h. |
| 2.4 g |
With palladium 10% on activated carbon; hydrogen; In ethyl acetate; at 80℃; under 760.051 Torr; for 12h; |
General procedure: To a mixture of 5a (2.13g, 10mmol) in dry ethyl acetate was added 10% Pd/C (0.5g), following the reaction mixture was stirred under hydrogen gas at 80C for 12h. The reaction mixture was concentrated in vacuo and the crude product was purified by column chromatography (dichloromethane/methanol 15:1 by volume) to give 4-morpholinyl aniline 6a (1.5g, yield 85%). |
|
With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; |
General procedure: A suspension of 1.01 g (5 mmol) 1-bromo-4-nitrobenzene, 1.5 g K2CO3, 0.59 mL (6 mmol) piperidine in 10 mL of DMF was heated to reflux overnight. Upon cooling, the reaction mixture was dilute with water, extracted with EA, and the organic layer was washed with water, followed by saturated NaCl aqueous solution, dried over anhydrous Na2SO4 and purified by flash chromatography (PE : EA = 50:1, 30:1) to afford 902 mg (87%) yellow solid. The solid was dissolved in methanol, 90 mg Pd-C (10%) was added and stirred under hydrogen overnight at room temperature and then filtered through Celite and concentratedin vacuo. The crude product was purified by flash chromatography (PEEA = 5:1) to afford 4l’ 0.706 g 99%. |
|
With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; |
A solution of 4-fluoronitrobenzene (7.05g, 50mmol), and K2CO3 (6.91g, 50 mmol) in DMSO (10mL) was stirred at room temperature for 0.5h. Subsequently, 1-methylpiperazine (8.3mL, 75mmol) was added dropwise, and stirred overnight. The mixture was then poured into ice-water, and the yellow precipitate collected by filtration to give 12 and dried. Compound 12 (2.2g, 10mmol) was dissolved in methanol (60mL), 10% Pd/C (0.3g) added, and stirred at room temperature under an atmosphere of H2 overnight. After completion of the reaction, the resulting mixture was filtered, and concentrated under vacuum to yield compound 13 as a purple solid. ESI-MS m/z: 192.3 (M+H)+, calcd for C11H17N3: 191.14. |
|
With 5%-palladium/activated carbon; hydrogen; |
General procedure: The reaction of 2,4-dichloro-5-nitropyrimidine withisopropylamine produced intermediate 2-chloro-N-isopropyl-5-nitropyrimidin-4-amine. 4-Fluoronitrobenzene reacted with1-methylpiperazine in DMSO yielded the intermediate <strong>[16155-03-6]1-methyl-4-(4-nitrophenyl)piperazine</strong> in the presence of K2CO3. The catalytichydrogenation of <strong>[16155-03-6]1-methyl-4-(4-nitrophenyl)piperazine</strong> with palladiumon carbon (Pd/C, 5%) quantificationally provided thedesired 4-(4-methylpiperazin-1-yl) aniline. Refluxing of the 2-chloro-N-isopropyl-5-nitropyrimidin-4-amine with 4-(4-methylpiperazin-1-yl)aniline in n-butanol yielded N4-isopropyl-N2-(4-(4-methylpiperazin-1-yl)phenyl)-5-nitropyrimidine-2,4-diamine,which was reduced to intermediate A1 with a good yield by catalytichydrogenation using Pd/C as a catalyst. Intermediates A wereprepared as these steps and used for the next step without furtherpurification. These processes were carried out as reported |
|
With hydrazine; In ethanol;Reflux; |
General procedure: 4-Nitrobenzyl bromide (46.3mmol) was dissolved in dichloromethane (100mL). The solution was added to the mixture of relative amine (47.0mmol) and triethylamine (70.3mmol) in dichloromethane (20ml). The reaction mixture was stirred at r. t. for 24 h and was extracted with dichloromethane (100ml×3). After removal of the solvent, the residue was crystallized from ethanol, giving yellow powder. Compounds 1 and 2 were used for further reaction without purification. To a suspension of compounds 1-2 (36.2mmol) in 95% ethanol (100ml), 85% NH2NH2·H2O (362mmol), 95% ethanol (100ml) and iron (III) oxide hydroxide (FeO(OH)/C, 2.0g) were added and heated to reflux. When TLC analysis showed complete conversion of the starting material, the reaction mixture was filtrate through Cellit and the filtrate was concentrated in vacuum. The crude product was purified by silica gel colum chromatography (DCM/MeOH) to yield the title compound (3 and 4) as white solid. The mixture of compound 4 (1eq, 18.5mmol), 4-Nitro-1H-pyrazole-3-acid (1.1equiv, 20.4mmol), EDC (1.2equiv, 22.2mmol), HOBT (1.2equiv, 22.2mmol) in DMF (50ml) was stirred for 24h. The ice water (100ml) was added to the reaction mixture. A large amount of yellow solid precipitation (compound 8) was acquired. Compound 8 was used without further purification. Compounds 8 was reduced by the same process as compound 4, and then the resulting compound 12 was purified by column chromatography on silica gel, eluted with the appropriate solvent. |
|
|
General procedure: To 3 g of the corresponding halonitrobenzene in a round bottomflask, 10 mL of dimethylsulfoxide was added and stirred for 5 min followedby the addition of 4.4 g (1.5 eq) of potassium carbonate and 4.5 g(2 eq) of N-methylpiperazine. The reaction was refluxed for 5 h,monitored by thin layer chromatography, and upon completion, thereaction mixture was quenched with water, and extracted with dichloromethane.It was dried over anhydrous magnesium sulfate, andevaporated under vacuum. The resulting crude compound was purifiedusing dichloromethane: methanol 90/10 to yield bright orange solid(59% yield) of the 2(4)-(4-methylpiperazine)nitrobenzene (3a and3b).25 Immediately after, the orange solids were dissolved in tetrahydrofuran(THF) under inert atmosphere. To this solution was added,0.1 eq of Pd(OAc)2 dissolved in THF, followed by a solution of degassedsolution of 0.5 eq potassium fluoride dissolved in minimum amount ofde-ionized water. The resulting reaction mixture was cooled and stirredat 0-5 C for 15-20 min followed by the drop wise addition of 0.5 eq ofpolymethylhydrosiloxane (PMHS). Reaction mixture turned black uponthe addition of PMHS and the TLC monitoring was required immediatelyfollowing the addition of PMHS. Reaction mixture wasstirred for 10 min at room temperature after the addition of PMHS. Thecrude compound was filtered on a Celite column, washed with water,and extracted with diethyl ether, and evaporated in vacuum, and purifiedby automated flash chromatography (hexanes: ethyl acetate: methanol60/20/20). The resulted product was obtained in 72-85% yield.3-(4-methylpiperazino)aniline (4c) and 4-(4-ethylpiperazino) aniline(4d) were used from commercial sources |
| 250 mg |
With iron(0); ammonia hydrochloride; In methanol; lithium hydroxide monohydrate; at 100℃; |
General procedure: The compound S1 (261 mg, 1.25 mmol), iron powder (210 mg, 4.76 mmol, 3 equiv.) and ammonium chloride (335 mg, 6.27 mmol, 5 equiv.) were dissolved in methanol : water (2 : 1, 15 mL). The reaction mixture was heated at 100 C overnight, cooled to RT, filtered through celite and the solvent was reduced under vacuum. The condensed mixture was extracted with DCM, washed with brine, dried with sodium sulfate and all solvent was evaporated to furnish the condensed residue, which was purified by flash chromatography (elution system - EA/Hexane = 1 : 1 ) to obtain the title compound (245 mg, 1.43 mmol). |
|
With 5%-palladium/activated carbon; hydrogen; In ethanol; lithium hydroxide monohydrate; at 20℃; |
General procedure: The substituted nitro compound 11 (1 equiv in a mixture of EtOH-H2O, 95:5, 20mL) was treated with 10% Pd-carbon (5% w/w). The reaction was subjected to hydrogenation under hydrogen gas at room temperature and the reaction was monitored by TLC. After completion of the reaction, the mixture was filtered through a Celite bed and concentrated in a vacuum to afford product 12. |
| 2.1 g |
With palladium 10% on activated carbon; hydrogen; In ethanol; at 20℃; for 12h; |
General procedure: PdeC (0.27 g, 10% m/m) wasadded to a solution of 8a (2.7 g, 13.0 mmol) in ethanol (20 ml) andhydrogenated for 12 h at room temperature. The resultant wasfiltered, washed with ethanol and concentrated. 2.2 g of 9a aspurple solid was obtained; yield: 94%. |
|
With FeO(OH)/C; hydrazine hydrate monohydrate; In ethanol; at 0 - 80℃; for 4h; |
General procedure: To a solution of 1a (5 g, 22.5 mmol) in 95% ethanol (100 mL) wasadded goethite (FeO(OH))/C (1.0 g) at room temperature. And a solution of 80% hydrazine hydrate (25 mL, 400 mmol) in 95% ethanol (50 mL) was added dropwise to the mixture at 0 C. The reaction mixture was stirred at 80 C for 4 h. The solvent was removed in vacuo to give 2a (3.8 g, yield 74.5%). MS m/z: 278.2 [M+H]+. |
|
With palladium 10% on activated carbon; hydrogen; In ethyl acetate; at 20℃; for 16h; |
Step B: 4-(4-Methylpiperazin-1-yl)aniline To a solution of <strong>[16155-03-6]1-methyl-4-(4-nitrophenyl)piperazine</strong> (5.00g, 22.60mmol) in ethyl acetate (80mL) was added 10% palladiumcarbon (2.00g), and it was replaced three times with a hydrogen balloon, stirred at room temperature for 16 hours. It was filtered over Celite, rinsed five times with dichloromethane and methanol (200mL), and spin-dried to give the title compound. 1H NMR (400MHz, DMSO-d6): δ=6.67 (d, J=8.8 Hz, 2H), 6.48 (d, J=8.6 Hz, 2H), 4.54 (s, 2H), 2.93-2.80 (m, 4H), 2.46-2.36 (m, 4H), 2.19 (s, 3H). |
|
With palladium on activated charcoal; hydrogen; In ethanol; at 40℃; for 6h; |
To a solution of 1-fluoro-4-nitrobenzene (1 equiv), 1-methylpiperazine (1.1 equiv), potassium carbonate (3 equiv) in N,N-dimethylformamide. The reaction mixture was stirred at 80C. Upon completion, the mixture was diluted with water and extracted with CH2Cl2. The organic phases were combined, washed with saline, dried over anhydrous Na2SO4 and evaporated to afford crude product 23. Intermediate 23 (1 equiv) was dissolved in ethanol, and Pd/C (0.1 equiv) was added. The flask was flushed with H2and stirred for 6h at 40C. The reaction mixture was filtered through a Celite pad and the filtrate was purified using chromatography to produce intermediate 24. MS (ESI) m/z(%): 192.3 [M+H]+. |
|
With ammonia hydrochloride; zinc powder; In lithium hydroxide monohydrate; at 20 - 70℃; for 1h;Inert atmosphere; |
General procedure: 11a-b (50 mmol), 2,4,5-trichloropyrimidine (50 mmol) andDIPEA (100 mmol) in acetonitrile, it was gradually heated to 80 Cand stirred for 10 h. The precipitate was filtrated and washed withwater, and the acquired production could directly be used withoutfurther purification. 13a-k was prepared according to the reportedprocess [20e22,24]. Ar atmosphere, a mixture of 13a-k (15 mmol),ammonium chloride (20 mmol) and zinc powder (120 mmol) inethanol/water (100/20 mL) was reacted at 70 C for 1 h. After thereaction, the solvent was distilled off and the residuewas dispersedin dichloromethane. The zinc powder was removed by filtration,and the organic layer was washed with dilute ammonia and driedover anhydrous sodium sulfate. The organicwas vacuum distilled to give the crude which was purified by silica-gel column separationto get the 14a-k. A mixture of 12a-b (5.0 mmol), 14a-k (5.0 mmol), and p-TsOH(7.5 mmol) in ethyl alcohol was gradually heated to 70 C andstirred for 3 h. After reaction, the solvent was vacuum distilled andthe residuum was dispersed with saturated sodium bicarbonate/ethyl acetate. The crude product was purified using flash chromatographywith dichloromethane/methanol as eluents.Ar atmosphere,15a-k (2.0 mmol) and hydroxylamine (40 mmol)in anhydrous methanol, sodium methoxide (25 mmol) in methanolsolution was slowly added to the mixture. After the addition, thereaction was allowed to warm room temperature and stirred for2 h. Adjusted pH to 6 with dilute hydrochloric acid, the inorganicsalt was removed by filtration and the rest of organic phase wasvacuum distilled to produce the 9~a k. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping