| 90% |
|
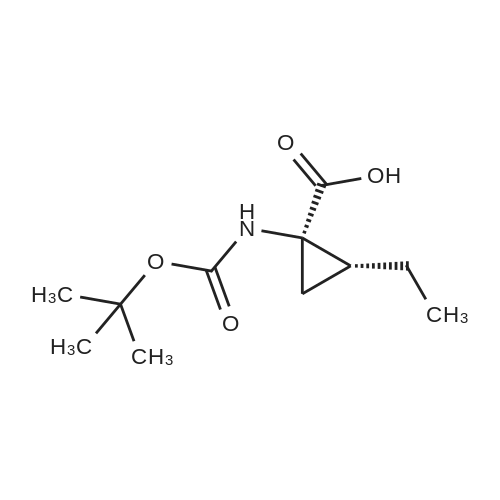
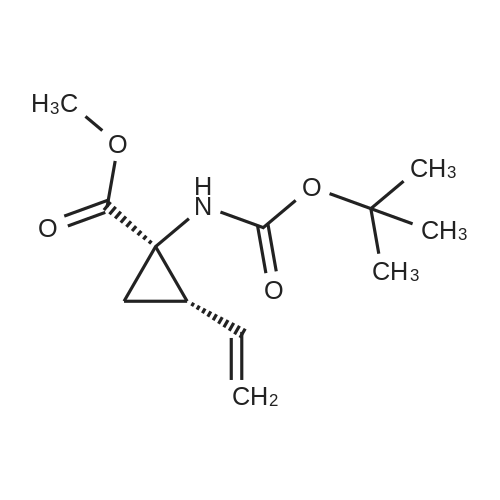
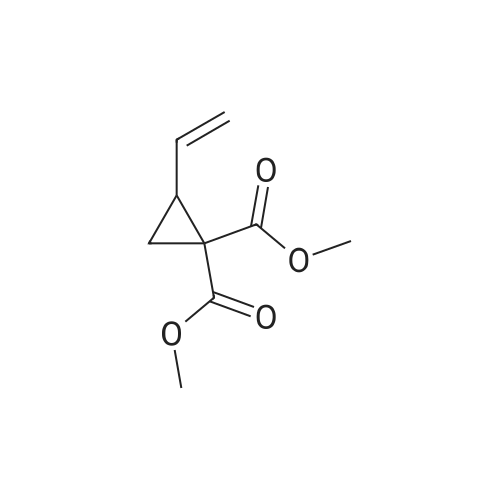
Example 1Synthesis of {4-Cyclopropanesulfonylaminocarbonyl-2,15-dioxo-18-[2-(4-trifluoromethyl-phenyl)-benzo[4,5]furo[3,2-d]pyrimidin-4-yloxy]-3,16-diaza-tricyclo[14.3.0.04,6]nonadec-14-yl}-carbamic acid cyclopentyl ester (Compound 1)Compound I-3 was first prepared from commercially available 1-t-butoxycarbonylamino-2-vinyl-cyclopropanecarboxylic acid ethyl ester via the route shown below:To a solution of 1-t-butoxycarbonylamino-2-vinyl-cyclopropanecarboxylic acid ethyl ester (0.34 g, 1.3 mmol) in THF (5 mL) and methanol (5 mL) was added a suspension of LiOH (0.13 g, 5.3 mmol) in water (1.4 mL). After being stirred overnight at room temperature, the reaction was quenched with 10percent HCl (2 mL) and the solvent was removed under vacuum. The resultant solid powder was washed with water (10 mL) to give compound I-1 (0.27 g, 90percent). MS m/z 249.9 (M++23); 1H NMR (CDCl3) delta 10.35 (brs, 1H), 5.84-5.71 (m, 1H), 5.29 (d, J=17.4 Hz, 1H), 5.12 (d, J=10.2 Hz, 1H), 2.23-2.14 (m, 1H), 1.87-1.65 (m, 1H), 1.58-1.41 (m, 1H), 1.43 (s, 9H).A solution of compound I-1 (0.52 g, 2.3 mmol), 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluoro-phosphate methanaminium (HATU, 1.74 g, 4.6 mmol), and 4-dimethylaminopyridine (1.39 g, 11.6 mmol) in CH2Cl2 (40 mL) was stirred at room temperature for 1 hour, followed by slow addition of cyclopropanesulfonamide (0.57 g, 4.7 mmol), diisopropylethylamine (1.81 mL, 14.0 mmol), and 1,8-diazabicyclo[5,4,0]undec-7-ene (1.80 g, 11.7 mmol) over 15 minutes. After the reaction mixture was stirred at room temperature overnight, the solvent was removed under vacuum. The residue was purified by silica gel column chromatography to give compound I-2 (0.51 g, 66percent). MS m/z 353.1 (M++423); 1H NMR (CDCl3) delta 9.75 (brs, 1H), 5.64-5.51 (m, 1H), 5.30 (d, J=17.4H), 5.16 (d, J=10.2 Hz, 1H), 2.95-2.89 (m, 1H), 2.19-2.10 (m, 1H), 1.93-1.88 (m, 1H), 1.47 (s, 9H), 1.46-1.38 (m, 1H), 1.32-1.23 (m, 2H), 1.15-1.00 (m, 2H).To a solution of compound I-2 (0.50 g, 1.5 mmol) in MeOH (8 mL) was added SOCl2 (0.26 g, 2.2 mmol) at room temperature. After the reaction mixture was refluxed for 1 hour, MeOH and SOCl2 was removed under vacuum. The residue was triturated from pentane and filtered to give intermediate I-3 as an off-white solid (0.32 g, 91percent). MS m/z (M++1); 1H NMR (CD3COD) delta 5.77-5.65 (m, 1H), 5.43 (d, J=17.4 Hz, 1H), 5.32 (d, J=10.2 Hz, 1H), 3.06-2.97 (m, 1H), 2.45 (dd, J=17.4 Hz, J=7.8, 1H), 2.16 (dd, J=8.0 Hz, J=7.8 Hz, 1H), 1.75 (dd, J=10.1 Hz, J=7.8 Hz, 1H), 1.32-0.86 (m, 4H).Compound 1 was prepared via the route shown below:A solution of 3-amino-benzofuran-2-carboxylic acid amide (1.00 g, 5.7 mmol) and pyridine (1 mL, 12.26 mmol) in THF (25 mL) was stirred at 0° C. for 10 min. To the resulting solution was slowly added 4-trifluoromethyl-benzoyl chloride (1.48 g, 7.1 mmol). Then the temperature was raised to room temperature and the mixture was stirred for 12 h. After the solvent was removed under reduced pressure, the resulting solid was collected, washed with water, and air-dried to yield I-4 (1.92 g, 96.0percent). MS: m/z 349.0 (M++1).To a suspension of I-4 (1.92 g, 5.5 mmol) and 2N NaOH (13 mL) in EtOH (25 mL) was heated at 85° C. for 12 h. After cooled, the mixture was acidified and then EtOH was removed. The resulting solid was collected, filtrated, washed with water, and dried to afford I-5 (1.71 g, 95.0percent). MS m/z 331 (M++1).A solution of I-5 (1.71 g, 5.2 mmol) and excess phosphorus oxychloride (POCl3) was refluxed for 2 hours. After cooled and thoroughly concentrated, the mixture was subjected to extraction with methylene chloride and 10percent sodium hydroxide. The organic layer was dried over MgSO4, concentrated, and crystallized from CH2Cl2 and n-hexane to give compound I-6 (1.49 g, 82percent). MS m/z 348.8, 350.9 (M++1); 1H NMR (CDCl3) delta 8.70 (d, 2H), 8.34 (d, 1H), 7.82-7.75 (m, 4H), 7.57 (ddd, 1H).To a suspension of boc-trans-4-hydroxy-L-proline (0.53 g, 2.3 mmol) in DMSO (25 mL) was added t-BuOK (0.82 g, 5.1 mmol) at 0° C. After the mixture was allowed to warm to room temperature and stirred for 1 hour, compound I-6 (0.81 g, 2.3 mmol) was added slowly at 10° C. Stirring was continued overnight. Iodomethane (1.02 g, 6.9 mmol) was added and the reaction mixture was stirred at room temperature for additional 30 minutes. The reaction mixture was neutralized to pH 6-7 by 10percent HCl aqueous solution and subjected to extraction with methylene chloride. The organic layer was dried over MgSO4, evaporated under vacuum, and purified by silica gel column chromatography to give compound I-7 (1.12 g, 86percent). MS m/z 557.8 (M++1); 1H NMR (CDCl3) delta 8.63 (d, 2H), 8.28 (d, 1H), 7.80-7.74 (m, 2H), 7.70 (d, 2H), 7.51 (ddd, 1H).To a solution of compound I-7 (1.13 g, 2.0 mmol) in MeOH (20 mL) was added SOCl2 (1.21 g, 9.8 mmol) at room temperature. The reaction mixture was refluxed for 1 hour, and MeOH and SOCl2 were removed. The residue was triturated in penta... |
| 90% |
|
Compound 1-3 was first prepared from commercially available 1 -t- butoxycarbonylamino-2-vinyl-cyclopropanecarboxylic acid ethyl ester via the route shown below:To a solution of l-i-butoxycarbonylamino-2-vinyl-cyclopropanecarboxylic acid ethyl ester (0.34 g, 1.3 mmol) in THF (5 mL) and methanol (5 mL) was added a suspension of LiOH (0.13 g, 5.3 mmol) in water (1.4 mL). After being stirred overnight at room temperature, the reaction was quenched with 10percent HCI (2 mL) and the solvent was removed under vacuum. The resultant solid powder was washed with water (10 mL) to give compound I-l (0.27 g, 90percent). MS m/z 249.9 (M++23); 1H NMR (CDC13) d 10.35 (brs, 1H), 5.84-5.71 (m, 1H), 5.29 (d, J = 17.4 Hz, 1H), 5.12 (d, J = 10.2 Hz, 1H), 2.23-2.14 (m, 1H), 1.87-1.65 (m, 1H), 1.58-1.41 (m, 1H), 1.43 (s, 9H). |
| 87% |
With lithium hydroxide; water; In tetrahydrofuran; methanol;Product distribution / selectivity; |
III. Preparation of P1'-P1 Intermediates 1a. Preparation of cyclopropanesulfonic acid (1-(R)-amino-2-(S)-vinyl-cyclopropanecarbonyl)amide HCl salt Step 1: Preparation of 1(R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid To a solution of 1(R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid ethyl ester (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature and quenched with 1N NaOH (15 mL) and water (20 mL). The resulting mixture was washed with ethyl acetate (20 mL), and the organic phase was extracted with 20 mL 0.5N NaOH. The combined aqueous phases were acidified with 1N HCl until pH 4 and extracted with ethyl acetate (3.x.40 mL). The combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated to yield the title compound as a white solid (2.62 g, 87percent). 1H NMR: (DMSO-d6) delta 1.22-1.26 (m, 1H), 1.37 (s, 9H), 1.50-1.52 (m, 1H), 2.05 (q, J=9 Hz, 1H), 5.04 (d, J=10 Hz, 1H), 5.22 (d, J=17 Hz, 1H), 5.64-5.71 (m, 1H), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s, 1H)); LC-MS (retention time: 1.67 minutes, method B), MS m/z 228 (M++H).; Example 7 To a solution of 1 (R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid ethyl ester (prepared by the procedure described in WO 03/099274, 3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature and quenched with 1N NaOH (15 mL) and water (20 mL). The resulting mixture was washed with ethyl acetate (20 mL), and the organic phase was extracted with 20 mL 0.5N NaOH. The combined aqueous phases were acidified with 1N HCl to pH 4 and extracted with ethyl acetate (3.x.40 mL). The combined organic extracts were washed with brine, dried (MgSO4), filtered, and concentrated to provide the desired compound as a white solid (2.62 g, 87percent). 1H NMR (DMSO-d6) delta 1.22-1.26 (m, 1H), 1.37 (s, 9H), 1.50-1.52 (m, 1H), 2.05 (q, J=9 Hz, 1H), 5.04 (d, J=10 Hz, 1H), 5.22 (d, J=17 Hz, 1H), 5.64-5.71 (m, 1H), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s, 1H); LC/MS (MH+, 228). |
| 87% |
|
To a solution of 1(R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid ethyl ester (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature and quenched with 1N NaOH (15 mL) and water (20 mL). The resulting mixture was washed with ethyl acetate (20 mL), and the organic phase was extracted with 20 mL 0.5N NaOH. The combined aqueous phases were acidified with 1N HCl to pH 4 and extracted with ethyl acetate (3*40 mL). The combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated to provide the desired compound as a white solid (2.62 g, 87percent). 1H-NMR: (DMSO-d6) delta 1.22-1.26 (m, 1H), 1.37 (s, 9H), 1.50-1.52 (m, 1H), 2.05 (q, J=9 Hz, 1H), 5.04 (d, J=10 Hz, 1H), 5.22 (d, J=17 Hz, 1H), 5.64-5.71 (m, 1H), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s, 1H)); LC-MS (retention time: 1.67 minutes, method B), MS m/z 228 (M++H). |
| 87% |
|
To a solution of 1(R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid ethyl ester (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature and quenched with 1N NaOH (15 mL) and water (20 mL). The resulting mixture was washed with ethyl acetate (20 mL), and the organic phase was extracted with 20 mL 0.5 N NaOH. The combined aqueous phases were acidified with 1N HCl until pH 4 and extracted with ethyl acetate (3.x.40 mL). The combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated to yield the title compound as a white solid (2.62 g, 87percent). 1H NMR: (DMSO-d6) delta 1.22-1.26 (m, 1H), 1.37 (s, 9H), 1.50-1.52 (m, 1H), 2.05 (q, J=9 Hz, 1H), 5.04 (d, J=10 Hz, 1H), 5.22 (d, J=17 Hz, 1H), 5.64-5.71 (m, 1H), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s, 1H)); MS m/z 228 (M++H). |
| 87% |
|
To a solution ofl (R)-tert-butoxycarbonylamino-2 (S)-vinyl- cyclopropanecarboxylic acid ethyl ester, the product of Step 6a (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspensionof LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature and quenched with IN NaOH (15mL) and water (20 mL). The resulting mixture was washed with EtOAc (20 mL), and the organic phase was extracted with 20 mL 0.5N NaOH. The combined aqueous phases were acidified with IN HCI until pH 4 and extracted with EtOAc (3 x 40mL). The combined organic extracts were washed with brine and dried(MgS04) to yield the title compound as a white solid (2.62 g, 87percent). 'H NMR :(DMSO-d6)b 1.22-1. 26 (m, 1H), 1.37 (s, 9H), 1.50-1. 52 (m, 1H), 2.05 (q, J=9 Hz,1H), 5.04 (d, J=10 Hz, 1H), 5.22 (d, J=17 Hz,1H), 5.64-5. 71(m, 1H), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s,1H)) ; LC-MS (retention time: 1.67 min, method B), MS7rl/z 228 (M++H). |
| 87% |
|
To a solution of 1(R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid ethyl ester (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature. To the mixture was added 1.0M NaOH (15 mL), water (20 mL) and ethyl acetate (20 mL). The mixture was shaken, the phases were separated, and the organic phase was again extracted with 20 mL 0.5M NaOH. The combined aqueous phases were acidified with 1.0M HCl until pH=4 and extracted with ethyl acetate (3.x.40 mL). The combined organic extracts were washed with brine, dried (MgSO4), and filtered to provide the desired product as a white solid (2.62 g, 87percent). 1H NMR: (DMSO-d6) delta1.22-1.26 (m, 1H), 1.37 (s, 9H), 1.50-1.52 (m, 1H), 2.05 (q, J=9 Hz, 1H), 5.04 (d, J=10 Hz, 1H), 5.22 (d, J=17 Hz, 1H), 5.64-5.71 (m, 1H), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s, 1H)); LC-MS MS m/z 228 (M++H). |
| 87% |
|
Step 1: Preparation of 1 (R)-tert-butoxycarbonylamino-2(S)-vinyl- cyclopropanecarboxylic acid; <n="66"/>To a solution of l(R)-tert-butoxycarbonylamino-2(S)-vinyl- cyclopropanecarboxylic acid ethyl ester (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature and quenched with IN NaOH (15 mL) and water (20 mL). The resulting mixture was washed with ethyl acetate (20 mL), and the organic phase was extracted with 20 mL 0.5N NaOH. The combined aqueous phases were acidified with IN HCl until pH 4 and extracted with ethyl acetate (3 x 4OmL). The combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated to yield the title compound as a white solid (2.62 g, 87percent). 1H NMR: (DMSO-d6) delta 1.22-1.26 (m, IH), 1.37 (s, 9H), 1.50-1.52 (m, IH), 2.05 (q, J=9 Hz, IH), 5.04 (d, J=IO Hz, IH), 5.22 (d, J=Il Hz, IH), 5.64- 5.71 (m, IH), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s, IH) ); MS m/z 228 (M++H). |
| 87% |
|
To a solution of 1(R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid ethyl ester (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature and quenched with 1N NaOH (15 mL) and water (20 mL). The resulting mixture was washed with ethyl acetate (20 mL), and the organic phase was extracted with 20 mL 0.5N NaOH. The combined aqueous phases were acidified with 1N HCl until pH 4 and extracted with ethyl acetate (3*40 mL). The combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated to yield the title compound as a white solid (2.62 g, 87percent). 1H NMR: (DMSO-d6) 1.22-1.26 (m, 1H), 1.37 (s, 9H), 1.50-1.52 (m, 1H), 2.05 (q, J=9 Hz, 1H), 5.04 (d, J=10 Hz, 1H), 5.22 (d, J=17 Hz, 1H), 5.64-5.71 (m, 1H), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s, 1H)); MS m/z 228 (M++H). |
| 87% |
|
Step 1: Preparation of 1(R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid To a solution of 1(R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid ethyl ester (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature and quenched with 1N NaOH (15 mL) and water (20 mL). The resulting mixture was washed with ethyl acetate (20 mL), and the organic phase was extracted with 20 mL 0.5N NaOH. The combined aqueous phases were acidified with 1N HCl until pH 4 and extracted with ethyl acetate (3*40 mL). The combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated to yield the title compound as a white solid (2.62 g, 87percent). 1H NMR: (DMSO-d6) delta 1.22-1.26 (m, 1H), 1.37 (s, 9H), 1.50-1.52 (m, 1H), 2.05 (q, J=9 Hz, 1H), 5.04 (d, J=10 Hz, 1H), 5.22 (d, J=17 Hz, 1H), 5.64-5.71 (m, 1H), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s, 1H)); MS m/z 228 (M++H). |
| 87% |
With lithium hydroxide; water; In tetrahydrofuran; methanol; at 20℃; |
To a solution of 1(R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid ethyl ester (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature and quenched with 1N NaOH (15 mL) and water (20 mL). The resulting mixture was washed with ethyl acetate (20 mL), and the organic phase was extracted with 20 mL 0.5N NaOH. The combined aqueous phases were acidified with 1N HCl until pH 4 and extracted with ethyl acetate (3.x.40 mL). The combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated to yield the title compound as a white solid (2.62 g, 87percent). 1H NMR: (DMSO-d6) 1.22-1.26 (m, 1H), 1.37 (s, 9H), 1.50-1.52 (m, 1H), 2.05 (q, J=9 Hz, 1H), 5.04 (d, J=10 Hz, 1H), 5.22 (d, J=17 Hz, 1H), 5.64-5.71 (m, 1H), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s, 1H)); MS m/z 228 (M++H). |
| 87% |
With lithium hydroxide; water; In tetrahydrofuran; methanol; at 20℃; |
To a solution of 1(R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid ethyl ester (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature and quenched with 1N NaOH (15 mL) and water (20 mL). The resulting mixture was washed with ethyl acetate (20 mL), and the organic phase was extracted with 20 mL 0.5N NaOH. The combined aqueous phases were acidified with 1N HCl until pH 4 and extracted with ethyl acetate (3.x.40 mL). The combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated to yield the title compound as a white solid (2.62 g, 87percent). 1H NMR: (DMSO-d6) |
| 87% |
|
III. Preparation of P1'-P1 Intermediates; 12. Preparation of P1-P1'; Step 1:; To a solution of 1(R)-tert-butoxycarbonylamino-2(S)-vinyl-cyclopropanecarboxylic acid ethyl ester (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature. To the mixture was added 10M NaOH (15 mL), water (20 mL) and ethyl acetate (20 mL). The mixture was shaken, the phases were separated, and the organic phase was again extracted with 20 mL 0.5M NaOH. The combined aqueous phases were acidified with 1.0M HCl until pH=4 and extracted with ethyl acetate (3.x.40 mL). The combined organic extracts were washed with brine, dried (MgSO4), and filtered to provide the desired product as a white solid (2.62 g, 87percent). 1H NMR: (DMSO-d6) delta1.22-1.26 (m, 1H), 1.37 (s, 9H), 1.50-1.52 (m, 1H), 2.05 (q, J=9 Hz, 1H), 5.04 (d, J=10 Hz, 1H), 5.22 (d, J=17 Hz, 1H), 5.64-5.71 (m, 1H), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s, 1H)); LC-MS MS m/z 228 (M++H). |
| 87% |
|
To a solution of l(R)-tert-butoxycarbonylamino-2(S)-vinyl- cyclopropanecarboxylic acid ethyl ester (3.28 g, 13.2 mmol) in THF (7 mL) and methanol (7 mL) was added a suspension of LiOH (1.27 g, 53.0 mmol) in water (14 mL). The mixture was stirred overnight at room temperature and quenched with IN NaOH (15 mL) and water (20 mL). The resulting mixture was washed with ethyl acetate (20 mL), and the organic phase was extracted with 20 mL 0.5N NaOH. The combined aqueous phases were acidified with IN HCl until pH 4 and extracted with ethyl acetate (3 x 4OmL). The combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated to yield the title compound as a white solid (2.62 g, 87percent). 1H NMR: (DMSO-d6) delta 1.22-1.26 (m, IH), 1.37 (s, 9H), 1.50-1.52 (m, IH), 2.05 (q, J=9 Hz, IH), 5.04 (d, J=IO Hz, IH), 5.22 (d, J=17 Hz, IH), 5.64- 5.71 (m, IH), 7.18, 7.53 (s, NH (rotamers), 12.4 (br s, IH) ); MS m/z 228 (M++H). |
| 71.44 g (54.0 g, 100%, corrected for residual solvent) |
With hydrogenchloride; lithium hydroxide; lithium hydroxide monohydrate; In tetrahydrofuran; cyclohexane; water; |
Stage 1a: (1R,2S)-1-(tert-butoxycarbonylamino)-2-vinyl-cyclopropane-1-carboxylic acid (452) Ethyl (1R,2S)-1-(tert-butoxycarbonylamino)-2-vinyl-cyclopropane-1-carboxylate (61 g, 0.239 mol, 1.0 eq.) and tetrahydrofuran (700 mL) were charged into a 2 L round bottom flask placed in ice/water bath. Lithium hydroxide monohydrate (30 g, 0.714 mol, 3.0 eq.) was dissolved in water (800 mL) and added slowly to the mixture. The reaction mixture was heated at 50° C. for 18 hours. Monitoring the reaction conversion by LCMS showed some residual starting material so lithium hydroxide (20 g, 0.476 mol, 2 eq.) was added. The reaction was stirred further for 5 hours and then stirred at room temperature for 2 days. Monitoring the reaction conversion by LCMS showed complete conversion. The reaction mixture was acidified to pH 3 by slow addition of 1M hydrochloric acid then extracted with ethyl acetate (4*900 mL). The organic extracts were pooled, washed with brine (600 mL), dried over sodium sulfate, filtered and concentrated to dryness. Cyclohexane (100 mL) was added to the dried crude material and concentrated to give 71.44 g (54.0 g, 100percent, corrected for residual solvent) of the title compound as a pale yellow solid which contained residual cyclohexane (24.5percent w/was calculated from 1H-NMR). The compound was used in the next step without further purification. 1H NMR (500 MHz, CHLOROFORM-d) delta ppm 5.79 (dt, J=17.01, 9.65 Hz, 1H) 5.27 (br. s., 1H) 5.30 (d, J=17.09 Hz, 1H) 5.14 (d, J=10.38 Hz, 1H) 2.20 (q, J=8.85 Hz, 1H) 1.70-1.90 (m, 1H) 1.52-1.63 (m, 1H) 1.45 (s, 9H) |
|
With lithium hydroxide; In tetrahydrofuran; methanol; water; at 20℃; for 1h; |
Intermediate C6: tert-butyl (1R,2S)-1-(cyclopropylsulfonylcarbamoyl)-2-vinylcyclopropylcarbamate Following the procedure in Scheme III, Intermediate C5 (6.0 g, 23.5 mmol) was added in a solution of LiOH (3.0 g, 73.5 mmol) in THF/MeOH/H2O (20 mL/20 mL/10 mL). After 1 h at r.t., the pH was adjusted to ?4 with 1N HCl, aqueous layer was separated, EA was added, and the mixture was extracted with EA twice. The combined organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to give the yellow oil, which was used directly in the next reaction without further purification. Then the intermediate (5.6 g, 24.2 mmol) and CDI (5.2 g, 31.5 mmol) were dissolved in THF (20 mL). After refluxed for 1 h, the mixture was cooled to r.t. and then a solution of cyclopropanesulfonamide (3.8 g, 31.5 mmol) in DCM (30 mL) was added followed by adding DBU (5.2 mg, 34.0 mmol). The mixture was stirred overnight at the room temperature, concentrated and extracted with EA twice. The combined organic layer was washed with brine, dried over Na2SO4, filtered, concentrated and purified by flash column chromatography to give the title compound C6 (2.8 g, 36.4percent) as white solid. 1H NMR (400 MHz, CDC3) delta 9.52 (s, 1H), 5.66-5.57 (m, 1H), 5.32 (d, J=13.2 Hz, 1H), 5.17 (dd, J=10.4, 0.8 Hz, 1H), 2.94-2.87 (m, 1H), 2.17 (q, J=8.4 Hz, 1H), 1.92-1.89 (m, 1H), 1.48 (s, 9H), 1.45-1.39 (m, 1H), 1.32-1.25 (m, 2H), 1.13-1.00 (m, 2H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping