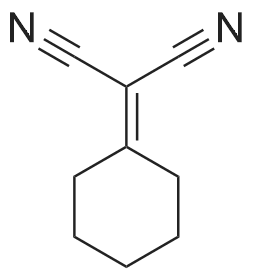
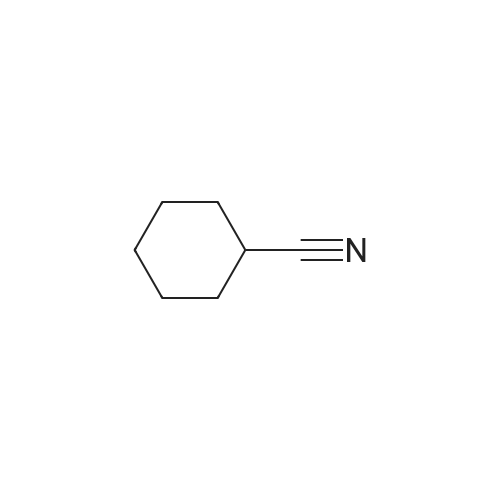
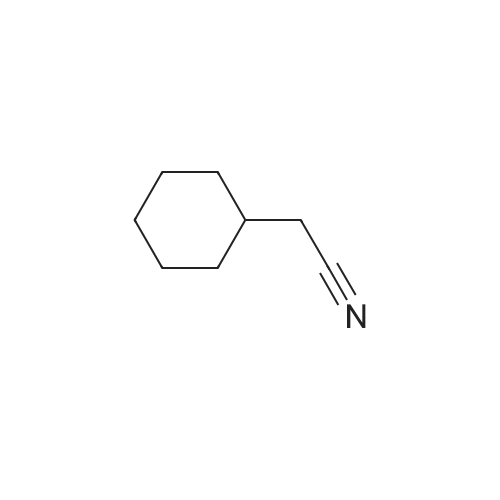
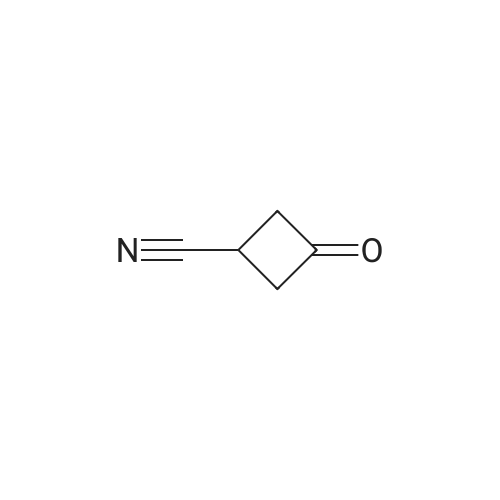
| 100% |
With water monomer; potassium hydroxide; In ethanol; |
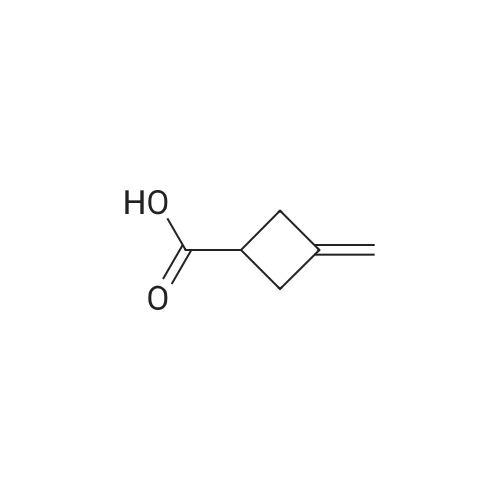
To a solution of 3- methylenecyclobutanecarbonitrile (1 g, 10.74 mmol) in EtOH (5.5 mL) and H2O (5.5 mL) was added KOH (2.410 g, 43.0 mmol). The reaction was refluxed overnight. The ethanol was removed under reduced pressure, The solution was cooled to 0 C and acidified to pHl with concentrated HC1. The mixture was extracted with EtOAc. The organic layer was washed with brine, dried and concentrated to yield E138A (1.204 g, 100%). NMR (500 MHz, CDCh) δ 4.90 - 4.75 (m, 2H), 3.22 - 3.08 (m, 1H), 3.08 - 2.86 (m, 4H). |
| 100% |
With water monomer; potassium hydroxide; In ethanol; at 80℃; for 3h; |
Dissolve 3-methylenecyclobutane-1-carbonitrile (3.1g, 33.7mmol) in 20mL ethanol,Add potassium hydroxide (7.6g, 135mmol) in water (20mL),React at 80C for 3 hours.After LCMS monitors that the reaction is complete, spin off the organic solvent,The aqueous phase was adjusted to pH 2 with concentrated hydrochloric acid, and extracted with ethyl acetate (50 mL×2).The combined organic phase was washed with saturated brine (50 mL), dried with anhydrous sodium sulfate,Filtered, spin-dried, and purified by column chromatography to obtain the product 23.1 (3.88 g, yield: 100%, colorless oil). |
| 98% |
With potassium hydroxide; In ethanol; at 20 - 90℃; |
To a stirred solution of KOH (10 g, 178 mmol) in water (15 mL) and EtOH (15 mL) was added 3 -methyl enecyclobutanecarbonitrile (3.92 g, 42 mmol) at room temperature for 10 min. The mixture was allowed to warm to 90 C and stirred at the same temperature for 3.5 h. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in water (10 mL) at 0 C. The mixture was acidified with 6 M aqueous HCl to pH 1 and extracted with DCM. The organic layer was dried over MgS04 and concentrated under reduced pressure to provide compound A141-1 (4.65 g, 98%) as a colorless oil. The product was used for next step without futher purification. |
| 97% |
With water monomer; potassium hydroxide; In ethanol; at 105℃; for 2.5h; |
To a solution of 3- methylidenecyclobutane-1-carbonitrile (6 g, 64.43 mmol, 1.00 eq.) in H20/EtOH (40/40 mL), was added potassium hydroxide (15 g, 267.33 mmol, 4.00 eq.) in several batches at 105C in 30 mm. The resulting solution was stirred for 2 hours at 105C. The resulting solution was dilutedwith water (200 mL) and the pH was adjusted to 2 with conc. hydrogen chloride aqueous (12M). The resulting solution was extracted with ethyl acetate (2x200 mL) and the organic layers combined. The resulting mixture was washed with brine (2x200 mL), dried over anhydrous sodium sulfate and concentrated under vacuum to give of 3-methylidenecyclobutane-1- carboxylic acid as yellow oil (7 g, 97%). |
| 97% |
With water monomer; potassium hydroxide; In ethanol; at 105℃; for 2.5h; |
Step la: 3-methyIenecyclobutane-l-carboxylic acid: To a solution of 3- methylidenecyclobutane-l -carbonitrile (6 g, 64.43 mmol, 1.00 eq.) in H20/EtOH (40/40 mL), was added potassium hydroxide (15 g, 267.33 mmol, 4.00 eq.) in several batches at 105C in 30 min. The resulting solution was stirred for 2 hours at 105 C. The resulting solution was diluted with water (200 mL) and the pH was adjusted to 2 with cone, hydrogen chloride aqueous (12 M). The resulting solution was extracted with ethyl acetate (2x200 mL) and the organic layers combined. The resulting mixture was washed with brine (2x200 mL), dried over anhydrous sodium sulfate and concentrated under vacuum to give of 3-methylidenecyclobutane-l - carboxylic acid as yellow oil (7 g, 97%). |
| 97% |
With water monomer; potassium hydroxide; In ethanol; at 105℃; for 2.5h; |
To a solution of 3- methylidenecyclobutane-l-carbonitrile (6 g, 64.43 mmol, 1.00 eq.) in H20/EtOH (40/40 mL), was added potassium hydroxide (15 g, 267.33 mmol, 4.00 eq.) in several batches at 105C in 30 min. The resulting solution was stirred for 2 hours at 105C. The resulting solution was diluted with water (200 mL) and the pH was adjusted to 2 with cone, hydrogen chloride aqueous (12 M). The resulting solution was extracted with ethyl acetate (2x200 mL) and the organic layers combined. The resulting mixture was washed with brine (2x200 mL), dried over anhydrous sodium sulfate and concentrated under vacuum to give of 3-methylidenecyclobutane-l- carboxylic acid as yellow oil (7 g, 97%). |
| 97% |
With water monomer; potassium hydroxide; In ethanol; at 105℃; for 2.5h; |
To a solution of 3- methylidenecyclobutane-l-carbonitrile (6 g, 64.43 mmol, 1.00 eq.) in H20/EtOH (40/40 mL), was added potassium hydroxide (15 g, 267.33 mmol, 4.00 eq.) in several batches at 105 C in 30 min. The resulting solution was stirred for 2 hours at 105C. The resulting solution was diluted with water (200 mL) and the pH was adjusted to 2 with cone, hydrogen chloride aqueous (12 M). The resulting solution was extracted with ethyl acetate (2x200 mL) and the organic layers combined. The resulting mixture was washed with brine (2x200 mL), dried over anhydrous sodium sulfate and concentrated under vacuum to give of 3- methylidenecyclobutane-l-carboxylic acid as yellow oil (7 g, 97%). |
| 96% |
With potassium hydroxide; In ethanol; water monomer; at 105℃; |
To a solution of 3-methylene-cyclobutanecarbonitrile (11.2 g, 120 mmol) in ethanol (30 mL) and water (30 mL) was added potassium hydroxide (33.7 g, 602 mmol). The mixture was stirred at 105 C overnight. After cooled down, it was concentrated. The aqueous residue was neutralized with concentrated hydrochloric acid and extracted with ethyl acetate (50 mL) for three times. The combined organic layers were washed withbrine (20 mL) twice, dried over Na2 SO4() and filtered. The filtrate was concentrated under reduced pressure to give the title compound (13.0 g, 96 % yield) as colorless oil.‘H NIVIR (400 1VIHz, DMSO-d6) 12.21 (s, 1H), 4.79-4.76 (m, 2H), 3.10-3.01 (m, 1H), 2.85 - 2.82 (m, 4H). |
| 95% |
With water monomer; potassium hydroxide; In ethanol;Reflux; |
To a solution of 3-methylenecyclobutanecarbonitrile (5.0 g, 53.7 mmol) in Ethanol (20 mL) was added aqueous KOH (35%) (34.4 g, 215 mmol) and the resulting mixture was refluxed overnight.The ethanol was removed under reduced pressure. The solution was then cooled below 10 C and acidified with concentrated HCI to pH = 5. The mixture was extracted with EtOAc (2x50 mL), the combined organic extracts were dried over anhydrous sodium sulfate and concentrated under vacuum to afford the title compound 3- methylenecyclobutanecarboxylic acid (6 g, 50.8 mmol, 95 % yield) as yellow oil. m/z: [M - H]" Calcd for C6H702 1 1 1 .1 ; Found 1 1 1 . |
| 94.5% |
With potassium hydroxide; In ethanol; water monomer; at 100℃; for 4h; |
3-methylenecyclobutane-1-carbonitrile (24 g, 258.0 mmol, 1.0 equiv) was dissolved in ethanol (180 mL) and water (180 mL). Potassium hydroxide (72.25 g, 1290.3 mmol, 5 equiv) was added and the reaction mixture was stirred at 100 C for 4 hours. The reaction mixture was concentrated. The residue was quenched with water, acidified by 1.0 N HCI aqueous solution to the pH 2 to 4 and extracted with EtOAc. The organic layer was washed with brine, dried over sodium sulfate and concentrated to afford product 3.1.57a (27.3 g, 94.5 % yield). LCMS (mlz): 111 [M+H]. 1H NMR (400 MHz, DMSO)6 12.25(s, 1H), 4.89-4.64(m, 2H), 3.06 (dt, J= 16.3, 8.0 Hz, 1H), 2.83(d, J8.1 Hz, 4H). |
| 94% |
With water monomer; potassium hydroxide; In ethanol; at 80℃; for 16h; |
Potassium hydroxide (61 g, 1087 mmol) was added to a solution of 3- methylenecyclobutanecarbonitrile (25 g, 268 mmol) in a mixture of ethanol (150 mL) and water (150 mL) at 27C. The reaction mixture was heated at 80 C for sixteen hours. On completion, the reaction mixture was concentrated under reduced pressure to remove the ethanol. Ice water (250 mL) was added to the residue and the mixture was acidified with concentrated hydrochloric acid (pH = ~1) (250 mL), extracted with ethyl acetate (500 mL, 2X), dried over sodium sulfate, filtered, and concentrated under reduced pressure to afford 3-methylenecyclobutanecarboxylic acid (30 g, 252 mmol, 94% yield) as a colorless liquid. 1H NMR (400 MHz, CD3SOCD3) δ 2.82 (d, J = 8 Hz, 4 H), 3.04 (p, J = 8 Hz, 1 H), 4.76 (p, J = 2 Hz, 2 H), 12.20 (br s, 1 H); LC-MS (LC- ES) M+H = 1 13. |
| 91% |
With water monomer; potassium hydroxide; In ethanol; at 100℃; for 2h; |
3- methylenecyclobutanecarbonitrile (10.0g, 107mmol) and potassium hydroxide (18.1g, 322mmol) was dissolved in ethanol (100 mL) and water (50mL), and reacted at 100C for 2 hours. 1N hydrochloric acid (120 mL) was added. Extracted with dichloromethane (30mLx3), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give 3-methylene cyclobutane carboxylic acid (11.0 g of, yellow oil). Yield: 91%. |
| 90% |
With water monomer; potassium hydroxide; In ethanol; at 105℃; for 4h; |
The solution of 3-methylenecyclobutanecarbonitrile 1 (27.9g, 0.3mol) in ethanol (200mL) and water (200mL) was treated with potassium hydroxide (84g, 1.5mol). The resulting mixture was stirred and heated to 105C for 4h. The ethanol was removed under reduced pressure. The residue was cooled to 0C, and the pH was adjusted to 1 ~2, then extracted with ethyl acetate, dried over sodium sulfate4, filtered and the solvent was removed. (30.2g, yield= 90%). |
| 77% |
With potassium hydroxide; In ethanol; water monomer; at 80℃; for 16h; |
To a solution of commercially available 3-methylenecyclobutanecarbonitrile (R-06b-1 , 10 g, 107 mmol) in EtOH/H20 (1 :1 , 200 ml.) was added KOH (24.1 g, 430 mmol) and the mixture was stirred at 80 C for 16 hours. Then, EtOH was removed under reduced pressure and then the solution was cooled to below 10 C and acidified with 1 M HCI to pH 1 . The mixture was extracted with EtOAc and the combined organic layer was dried over anhydrous Na2S04,filtered and concentrated to obtain R-06b-2 (9.3 g, 77%) as yellow oilsolid. This compound was used in the next step without further characterization. |
| 68% |
|
Reference Example 1 3-methylenecyclobutanecarboxylic acid 3-Methylenecyclobutanecarbonitrile (CAS# 15760-35-7, 33.84 g, 0.368 mole), sodium hydroxide (38 g, 0.95 mole) and water (50 mL) were mixed and heated at 110 C. for 30 min to give a two-phase mixture. Tetrabutylammonium hydroxide (40 wt. % in water, 0.7 mL) was added and the organic phase became brown. Within 10 minutes, gas was evolved, and the two phases became well mixed to give a light brown homogeneous solution. Heating was continued at 110 C. for two days. The mixture was cooled to room temperature and then to 0 C. Concentrated hydrochloric acid (95 mL) was added very slowly to adjust the pH to 0.5-1.0. A white slurry was formed, which was extracted with ether (500 mL*2). The organic layer was dried (over Na2SO4), and concentrated to give an oil. This oil was distilled and collected at 108 C.-110 C. to give a colorless liquid (27.74 g, 68% yield). 1H NMR (300 MHz, CDCl3) δ ppm 4.77-4.88 (m, 2H), 3.10-3.24 (m, 1H), 2.86-3.09 (m, 4H). |
| 9% |
With water monomer; potassium hydroxide; In ethanol; for 16h;Reflux; |
2-((frans)-4-(Dibenzylamino)cyclohexyl)-1 ,1 ,1 -trifluoropropan-2-ol (Intermediate 20B) (0.27 g, 0.69 mmol) and 20 wt % palladium hydroxide on carbon (0.097 g, 0.14 mmol) were stirred in ethanol (6 mL) and purged with hydrogen via balloon (3X) before stirring under a hydrogen atmosphere at room temperature overnight. The reaction was filtered through a pad of Celite and rinsed with ethanol. The filtrate was concentrated under reduced pressure to give the title compound as an oil (136 mg, 93%). 1H NMR (CD3OD) δ 1 .02-1 .28 (m, 7 H), 1 .51 - 1 .61 (m, 1 H), 1 .79-2.00 (m, 4 H), 2.51 -2.62 (m, 1 H); LC-MS (LC-ES) M+H = 212. |
|
|
To a solution of 3- methylenecyclobutanecarbonitrile (10.0 g, 107.4 mmol) in ethanol (100 mL) and water (100 mL) was added potassium hydroxide (28.0 g, 430 mmol, 85% pure); the resulting mixture was refluxed for 8 h. Ethanol was removed under reduced pressure, then the solution was cooled to 0 C and acidified with conc. HC1 to pH = 1. The mixture was extracted with diethyl ether (4 x 100 mL). The combined organic phases were dried over anhydrous sodium sulfate. Concentration in vacuo afforded the desired product as a colorless oil; 1H NMR (CDC13, 400 MHz) 8 2.91-3. 18 (m, 4H), 3.14-3. 22 (m, 1H), 4.83 (m, 2H); 13C NMR (CDC13, 100 MHz) 8 32.95, 35.30, 107.14, 143.77, 181. 02 ppm |
|
|
To a solution of 3-methylenecyclobutanecarbonitrile (10.0 g, 107.4 mmol) in ethanol (100 mL) and water (100 mL) was added potassium hydroxide (28.0 g, 430 mmol, 85% pure); the resulting mixture was refluxed for 8 h. Ethanol was removed under reduced pressure, then the solution was cooled to 0 C. and acidified with conc. HCl to pH=1. The mixture was extracted with diethyl ether (4*100 mL). The combined organic phases were dried over anhydrous sodium sulfate. Concentration in vacuo afforded the desired product as a colorless oil; 1H NMR (CDCl3, 400 MHz) δ 2.91-3.18 (m, 4H), 3.14-3.22 (m, 1H), 4.83 (m, 2H); 3C NMR (CDCl3, 100 MHz) δ 32.95, 35.30, 107.14, 143.77, 181.02 ppm. |
|
|
To a solution of 3-methylenecyclobutanecarbonitrile (100.0 g, 1.042 mol) in ethanol (1.00 L) and water (1.00 L) was added potassium hydroxide (230.0 g, 4.2 mol). The resulting mixture was heated at reflux for 7 hr then the EtOH was removed in vacuo and the solution was cooled to 0 C. and acidified with (300.0 mL) of conc. HCl to pH=1. The mixture was extracted with diethyl ether (4×1 L) and the combined organic phases were dried over sodium sulfate, filtered and concentrated in vacuo to yield desired product. 1H NMR (400 MHz, CDCl3) δ ppm 2.64-3.44 (m, 5H), 4.60-4.98 (m, 2H) and 10.64 (br. s., 1H). |
|
|
3-Methylene-cyclobutane carboxylic acid To a stirring solution of KOH (70.0 g, 1.25 mol) in EtOH/H2O (500 mL,1 :1 v/v) was added 3-methylenecyclobutane carbonitrile (25.0 g, 0.26 mol) and the reaction mixture was refluxed for 6 h. The reaction progress was monitored by TLC and, upon completion, the mixture was cooled and acidified to pH 3-4 with HCl. The ethanol was evaporated, and the remaining aqueous layer was extracted with Et2O (200 mL). The organic layer was washed with water (2 x 20 mL), brine (30 ml), dried overNa2SO4, filtered and concentrated to dryness to yield 3-methylene-cyclobutane carboxylic acid, which was carried through to the next step without further purification:1H NMR (250 MHz, CDCl3) δ 10.75 (bs, 1 H), 4.80 (s, 2 H), 2.85-3.26 (m, 5 H). |
|
|
To a solution of 3-methylenecyclobutanecarbonitrile (100.0 g, 1.042 mol) in ethanol (1.00 L) and water (1.00 L) was added potassium hydroxide (230.0 g, 4.2 mol). The resulting mixture was heated at reflux for 7 hr then the EtOH was removed in vacuo and the solution was cooled to 0 C. and acidified with (300.0 mL) of conc. HCl to pH=1. The mixture was extracted with diethyl ether (4×1 L) and the combined organic phases were dried over sodium sulfate, filtered and concentrated in vacuo to yield desired product. 1H NMR (400 MHz, CDCl3) δ ppm 2.64-3.44 (m, 5H), 4.60-4.98 (m, 2H) and 10.64 (br.s., 1H). |
|
|
To a stirring solution of KOH (70.0 g, 1.25 mol) in EtOH/H2O (500 niL, 1:1 v/v) was added 3-methylenecyclobutane carbonitrile (25.0 g, 0.26 mol) and the reaction mixture was refluxed for 6 h. The reaction progress was monitored by TLC and, upon completion, the mixture was cooled and acidified to pH 3-4 with HCl. The ethanol was evaporated, and the remaining aqueous layer was extracted with Et2O (200 mL). The organic layer was washed with water (2 x 20 mL), brine (30 ml), dried over Na2SO4, filtered and concentrated to dryness to yield 3-methylene-cyclobutane carboxylic acid, which was carried through to the next step without further purification: 1H NMR (250 MHz, CDCl3) δ 10.75 (bs, 1 H), 4.80 (s, 2 H), 2.85-3.26 (m, 5 H). |
|
|
To a stirring solution of KOH (70.0 g, 1.25 mol) in EtOH/H20 (500 mL, 1 : 1 v/v) was added 3-methylenecyclobutane carbonitrile (25.0 g, 0.26 mol) and the reaction mixture was refluxed for 6 h. The reaction progress was monitored by TLC and, upon completion, the mixture was cooled and acidified to pH 3-4 with HCl. The ethanol was evaporated, and the remaining aqueous layer was extracted with Et20 (200 mL). The organic layer was washed with water (2 x 20 mL), brine (30 ml), dried over Na2S04, filtered and concentrated to dryness to yield 3-methylene-cyclobutane carboxylic acid, which was carried through to the next step without further purification: 1H NMR (250 MHz, CDC13) δ 10.75 (bs, 1 H), 4.80 (s, 2 H), 2.85-3.26 (m, 5 H). |
|
|
To a stirring solution of KOH (70.0 g, 1.25 mol) in EtOH/H20 (500 mL, 1:1 v/v) was added 3-methylenecyclobutane carbonitrile (25.0 g, 0.26 mol) and the reaction mixture was refluxed for 6 h. The reaction progress was monitored by TLC and, upon completion, the mixture was cooled and acidified to pH 3-4 with HCl. The ethanol was evaporated, and the remaining aqueous layer was extracted with Et20 (200 mL). The organic layer was washed with water (2 x 20 mL), brine (30 ml), dried over Na2S04, filtered and concentrated to dryness to yield 3-methylene-cyclobutane carboxylic acid, which was carried through to the next step without further purification: 1H NMR (250 MHz, CDC13) δ 10.75 (bs, 1 H), 4.80 (s, 2 H), 2.85-3.26 (m, 5 H). |
|
With water monomer; potassium hydroxide; In ethanol; for 6h;Reflux; |
To a stirring solution of KOH (70.0 g, 1.25 mol) in EtOH/H20 (500 mL,1:1 v/v) was added 3-methylenecyclobutane carbonitrile (25.0 g, 0.26 mol) and the reaction mixture was refluxed for 6 h. The reaction progress was monitored by TLC and, upon completion, the mixture was cooled and acidified to pH 3-4 with HCl. The ethanol was evaporated, and the remaining aqueous layer was extracted with Et20 (200 mL). The organic layer was washed with water (2 x 20 mL), brine (30 ml), dried over Na2S04, filtered and concentrated to dryness to yield 3-methylene-cyclobutane carboxylic acid, which was carried through to the next step without further purification: 1H NMR (250 MHz, CDCl3) δ 10.75 (bs, 1 H), 4.80 (s, 2 H), 2.85-3.26 (m, 5 H). |
|
With water monomer; potassium hydroxide; In ethanol; for 6h;Reflux; |
To a stirring solution of KOH (70.0 g, 1.25 mol) in EtOH/H20 (500 mL, 1:1 v/v) was added 3-methylenecyclobutane carbonitrile (25.0 g, 0.26 mol) and the reaction mixture was refluxed for 6 h. The reaction progress was monitored by TLC and, upon completion, the mixture was cooled and acidified to pH 3-4 with HCl. The ethanol was evaporated, and the remaining aqueous layer was extracted with Et20 (200 mL). The organic layer was washed with water (2 x 20 mL), brine (30 ml), dried over Na2SC filtered and concentrated to dryness to yield 3-methylene-cyclobutane carboxylic acid, which was carried through to the next step without further purification: ? NMR (250 MHz, CDC13) δ 10.75 (bs, 1 H), 4.80 (s, 2 H), 2.85-3.26 (m, 5 H). |
|
With potassium hydroxide; In ethanol; water monomer; at 100℃; for 2h; |
Step 1. 3-Methylenecyclobutanecarboxylic acid Into a round bottom flask equipped with a condenser was added 3-methylenecyclobutanecarbonitrile (BePharma, 10.0 g, 0.107 mol). To the flask was added a solution of potassium hydroxide (24.1 g, 0.365 mol) in ethanol (112 mL) and water (88 mL) and the mixture was heated at 100 C. After about 2 hours, ammonia evolution ceased and the solvent was evaporated to dryness under reduced pressure. The solids were dissolved in water (75 mL), cooled in an ice-bath, and acidified to pH of about 1 with concentrated hydrochloric acid. The resulting upper layer was extracted with dichloromethane twice. The organic layers were combined and dried over anhydrous magnesium sulfate. Removal of the organic solvents gave the desired crude product (11.8 g, 97.67%). |
|
With water monomer; potassium hydroxide; In ethanol; for 2h;Reflux; |
3-Methylenecyclobutanecarbonitrile (244 g; 2.62 mol) was added to a solution of potassium hydroxide (85 wt-% pellets; 750 g; 11.4 mol) in a mixture of water (2.7 L) and EtOH (2,7 L). The mixture was heated to obtain a gentle reflux. The reaction was complete, after approximately 2 h, when a moist pH paper did not color blue at the top of the reflux condenser, i.e. ammonia evolution had ceased. The mixture was cooled and was subsequently extracted with toluene (2 x 0.5 L) and Et20 (4 x 0.5 L). The combined organic phases were dried overNa2SC>4. After removal of the drying agent, the resulting solution was concentrated under reduced pressure yielding an oily residue. 13C NMR (CDC13): δ 179.3; 146.2; 107.5; 36.7; 34.8. |
|
With potassium hydroxide; In ethanol; water monomer; for 2h;Reflux; |
Compound I- 10 was bought from a commercial source and the hydrolysis of I- 10 was carried out following a literature procedure (J. Am. Chem. Soc. 1958, 80, 5507). To a solution of 3-methylenecyclobutanecarbonitrile I- 10 (1 equivalent) in aqueous EtOH (50%) was added KOH (4 equivalents) and the homogeneous mixture was heated to reflux for 2 h. Upon cooling, all the volatile materials were evaporated and the solid was suspended in water. The pH of the solution was adjusted to 2 by the addition of IN HCl and the desired compound was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous MgS04 and evaporated to obtain 3-methylenecyclobutanecarboxylic acid in quantitative yield. This material was carried forward directly for the next step. To a suspension of LiAlH4 (1.5 equivalents) in THF was added 3-methylene- cyclobutanecarboxylic acid (in THF) (1 equivalent) slowly at 0 C. The cooling bath was removed and the reaction mixture was warmed to room temperature and stirred for 3 h. Following a Fischer workup, the desired compound 1-11 was obtained as colorless oil in 89% yield. This material was used for the next step without any purification. |
|
With water monomer; potassium hydroxide; In ethanol; for 2h;Reflux; |
Compound 1-10 was bought from a commercial source and the hydrolysis of I- 10 was carried out following a literature procedure (J. Am. Chem. Soc. 1958, 80, 5507). To a solution of 3-methylenecyclobutanecarbonitrile 1-10 (1 equivalent) in aqueous EtOH (50%) was added KOH (4 equivalents) and the homogeneous mixture was heated to reflux for 2 h. Upon cooling, all the volatile materials were evaporated and the solid was suspended in water. The pH of the solution was adjusted to 2 by the addition of IN HC1 and the desired compound was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous MgS04 and evaporated to obtain 3-methylenecyclobutanecarboxylic acid in quantitative yield. This material was carried forward directly for the next step. |
| 6.01 g |
With water monomer; potassium hydroxide; In ethanol;Reflux; |
Description 653-Methylenecyclobutanecarboxylic acid (D65) To a solution of 3-methylenecyclobutanecarbonitrile (5.0 g) in ethanol (20 mL) was added KOH aqueous solution (35%, 34.4 g) and the resulting mixture was heated to reflux overnight. The ethanol was removed under reduced pressure. The residue was cooled to below 10C and acidifiedwith concentrated HC1 to pH = 5. The mixture was extracted with EtOAc (2x50 mL). The combined organic extracts were dried over anhydrous sodium sulfate and concentrated under vacuum to afford the title compound (6.01 g) as yellow oil. MS (ESI): C6H802 requires 112; found 111 [M-Hf. |
|
With ethanol; potassium hydroxide;Reflux; |
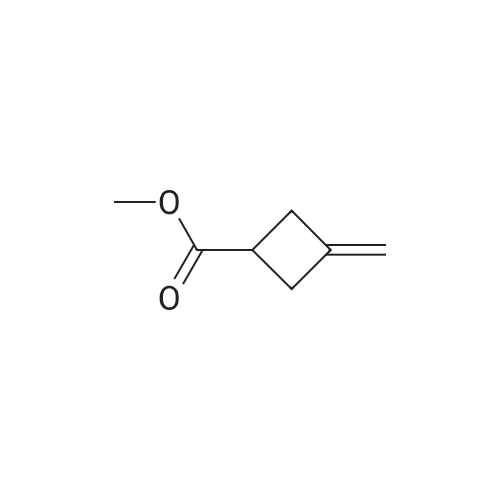
Preparation of reagent R-1 1 a: ethyl 3-methylenecvclobutanecarboxylate To a solution of commercially available 3-methylenecyclobutanecarbonitrile (10.7 g, 1 15 mmol) in EtOH (70 mL) was added KOH (25.2 g, 450 mmol), then the mixture was stirred at reflux overnight. The resulting mixture was stirred at reflux overnight until TLC showed the starting material was consumed completely, the solvent was evaporated and water was added. 1 N HCI was added to bring pH to ~3 and extracted with EtOAc, the organic layer was washed with brine, dried over anhydrous Na2SO4, concentrated to give the crude product (12.3 g,~95%). The crude product (12.3 g, 1 10 mmol) was dissolved in DMF (120 mL). Then Etl (21 .5 g, 138 mmol) and K2CO3 (31 .7 g, 230 mmol) were added to this solution. The mixture was stirred at r.t. for 8 h. Then water was added and extracted with EtOAc, the organic layer was washed with brine, dried over anhydrous Na2SO4, concentrated to give a crude which was purified by column to give the reagent R-1 1 a (13.6 g, 88% yield) as a pale yellow oil. GC-MS (M): 140 calc. for C8H12O2: 140.08. |
|
|
Example 17; Preparation of tert-butyl-3 -oxocyclobutyl carbamate; A solution of 70 g of KOH in 500 ml of mixture EtOH/H2O (1/1, v/v) was added to a 2 L round bottom flask equipped with magnetic stir bar and condenser followed by 3 -methyl enecyclobutane carbonitrile (Maybrige) (25 g, 0.26 mol). The reaction mixture was refluxed with stirring in an oil bath for 5-6 hours. The reaction was monitored for completion by TLC. Upon completion of reaction, the mixture was cooled and acidified with HCl to a pH of 3-4. Ethanol was evaporated, and the remaining aqueous layer was extracted with 200 mL of Et2O. Combined organics were washed with water (2 x 20 mL) followed by brine (once by 30 ml). Organics were dried over Na2SO4, filtered and evaporated. The resulting product, 3- methylenecyclobutane carboxylic acid (shown above), was used without further purification in the next step. |
|
|
Example 46; 6'-(Methyl-(l-hydroxy-3-amino-cyclobutyl)-l-(4-amino-2(S)-hydroxy-butyryl)- sisomicin3-Methylene-cyclobutane carboxylic acidTo a stirring solution of KOH (70.0 g, 1.25 mol) in EtOH/H2O (500 mL, 1 :1 v/v) was added 3 -methyl enecyclobutane carbonitrile (25.0 g, 0.26 mol) and the reaction mixture was refluxed for 6 h. The reaction progress was monitored by TLC and, upon completion, the mixture was cooled and acidified to pH 3-4 with HCl. The ethanol was evaporated, and the remaining aqueous layer was extracted with Et2O (200 mL). The organic layer was washed with water (2 x 20 mL), brine (30 ml), dried over Na2SO4, filtered and concentrated to dryness to yield 3-methylene-cyclobutane carboxylic acid, which was carried through to the next step without further purification: 1H NMR (250 MHz, CDCl3) δ 10.75 (bs, 1 H), 4.80 (s, 2 H), 2.85-3.26 (m, 5 H). |
|
|
3-Methylene-cyclobutane carboxylic acid To a stirring solution of KOH (70.0 g, 1.25 mol) in EtOH/H2O (500 mL,1 :1 v/v) was added 3-methylenecyclobutane carbonitrile (25.0 g, 0.26 mol) and the reaction mixture was refluxed for 6 h. The reaction progress was monitored by TLC and, upon completion, the mixture was cooled and acidified to pH 3-4 with HCl. The ethanol was evaporated, and the remaining aqueous layer was extracted with Et2O (200 mL). The organic layer was washed with water (2 x 20 mL), brine (30 ml), dried overNa2SO4, filtered and concentrated to dryness to yield 3-methylene-cyclobutane carboxylic acid, which was carried through to the next step without further purification:1H NMR (250 MHz, CDCl3) δ 10.75 (bs, 1 H), 4.80 (s, 2 H), 2.85-3.26 (m, 5 H). |
|
|
3-Methylene-cyclobutane carboxylic acidTo a stirring solution of KOH (70.0 g, 1.25 mol) in EtOH/H2O (500 mL, 1:1 v/v) was added 3-methylenecyclobutane carbonitrile (25.0 g, 0.26 mol) and the reaction mixture was refluxed for 6 h. The reaction progress was monitored by TLC and, upon completion, the mixture was cooled and acidified to pH 3-4 with HCl. The ethanol was evaporated, and the remaining aqueous layer was extracted with Et2O (200 mL). The organic layer was washed with water (2 x 20 mL), brine (30 ml), dried over Na2SO4, filtered and concentrated to dryness to yield 3-methylene-cyclobutane carboxylic acid, which was carried through to the next step without further purification:1H NMR (250 MHz, CDCl3) δ 10.75 (bs, 1 H), 4.80 (s, 2 H), 2.85-3.26 (m, 5 H).N-Boc-S-Methylene-cyclobutanainine |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +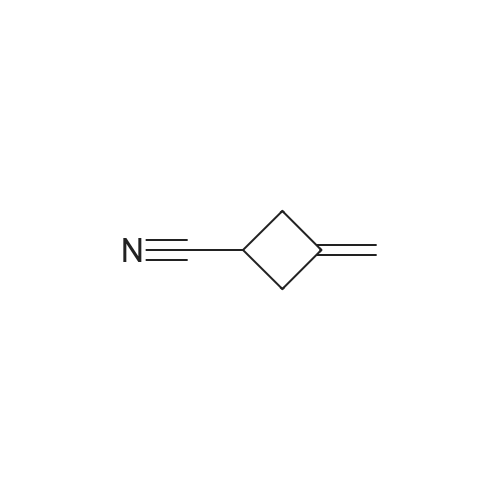

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping