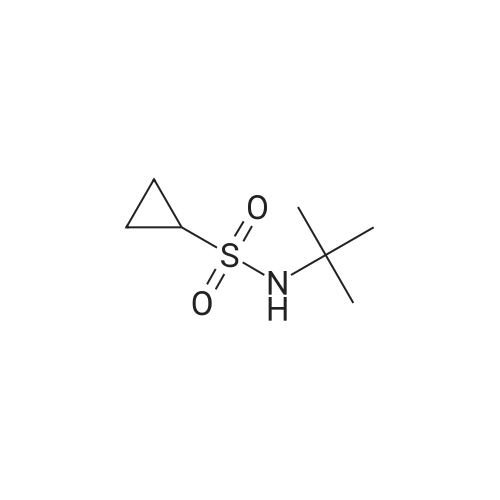
| 92% |
|
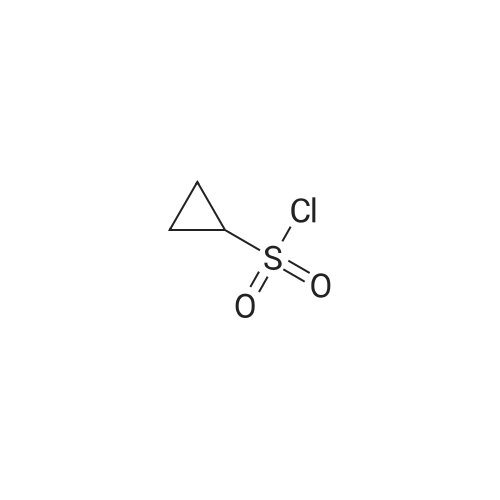
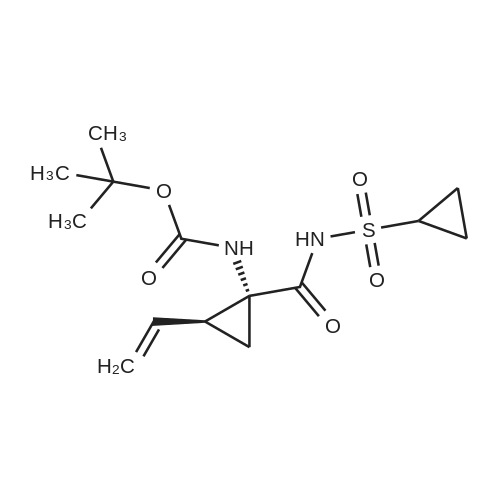
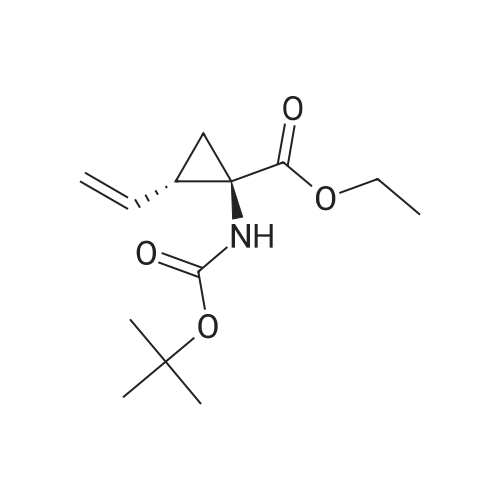
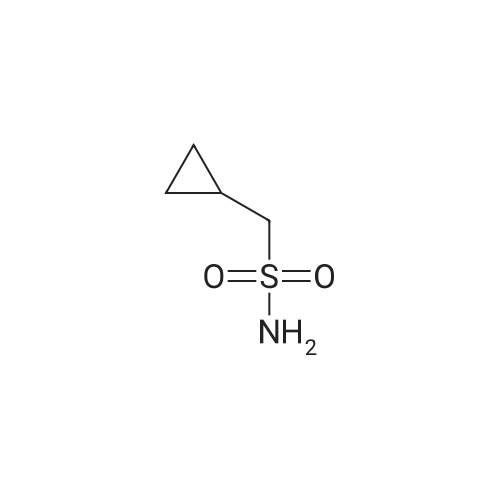
Step 2: Preparation of cyclopropanesulfonic acid (l-(R)-tert-butoxycarbonylamino- 2-(S)-vinylcyclopropanecarbonyl)-amide; A solution of the product of Step 1 (2.62 g, 11.5 mmol) and CDI (2.43 g, 15.0 mmol) in THF (40 mL) was heated at reflux for 50 minutes under nitrogen. The solution was cooled to room temperature and transferred by cannula to a solution of cyclopropylsulfonamide (1.82 g, 15.0 mmol) in THF (10 mL). To the resulting solution was added DBU (2.40 mL, 16.1 mmol) and stirring was continued for 20 hours. The mixture was quenched with IN HCl to pH 1 and THF was concentrated in vacuo. The suspension was extracted with ethyl acetate (2 x 50 mL) and the combined organic extracts were dried (Na2SO4), filtered, and concentrated. Purification by recystallization from hexanes-ethyl acetate (1: 1) afforded the title compound (2.4 g) as a white solid. The mother liquor was purified by a Biotage 4OS column (eluted 9percent acetone in dichloromethane) to give a second batch of the title compound (1.1 g). Both batches were combined (total yield 92percent). 1H NMR <n="67"/>(DMSO-d6) delta 0.96-1.10 (m, 4H), 1.22 (dd, J=S.5, 9.5 Hz, IH), 1.39 (s, 9H), 1.70 (t, J=5.5 Hz, IH), 2.19-2.24 (m, IH), 2.90 (m, IH), 5.08 (d, J=IO Hz, IH), 5.23 (d, J=Yl Hz, IH), 5.45 (m, IH), 6.85, 7.22 (s, NH (rotamers); MS mk 331 (M++H). |
| 92% |
|
A solution of the product of Step 1 (2.62 g, 11.5 mmol) and CDI (2.43 g, 15.0 mmol) in THF (40 mL) was heated at reflux for 50 minutes under nitrogen. The solution was cooled to room temperature and transferred by cannula to a solution of cyclopropylsulfonamide (1.82 g, 15.0 mmol) in THF (10 mL). To the resulting solution was added DBU (2.40 mL, 16.1 mmol) and stirring was continued for 20 hours. The mixture was quenched with 1N HCl to pH 1 and THF was concentrated in vacuo. The suspension was extracted with ethyl acetate (2*50 mL) and the combined organic extracts were dried (Na2SO4), filtered, and concentrated. Purification by recystallization from hexanes-ethyl acetate (1:1) afforded the title compound (2.4 g) as a white solid. The mother liquor was purified by a Biotage 40S column (eluted 9percent acetone in dichloromethane) to give a second batch of the title compound (1.1 g). Both batches were combined (total yield 92percent). 1H NMR (DMSO-d6) delta 0.96-1.10 (m, 4H), 1.22 (dd, J=5.5, 9.5 Hz, 1H), 1.39 (s, 9H), 1.70 (t, J=5.5 Hz, 1H), 2.19-2.24 (m, 1H), 2.90 (m, 1H), 5.08 (d, J=10 Hz, 1H), 5.23 (d, J=17 Hz, 1H), 5.45 (m, 1H), 6.85, 7.22 (s, NH (rotamers); MS m/z 331 (M++H). |
| 92% |
|
A solution of <strong>[159622-10-3](1R,2S)-1-((tert-butoxycarbonyl)amino)-2-vinylcyclopropanecarboxylic acid</strong> (2.62 g, 11.5 mmol) and CDI (2.43 g, 15.0 mmol) in THF (40 mL) was heated at reflux for 50 min under nitrogen. The solution was cooled to room temperature and transferred by cannula to a solution of cyclopropylsulfonamide (1.82 g, 15.0 mmol) in THF (10 mL). To the resulting solution was added DBU (2.40 mL, 16.1 mmol) and stirring was continued for 20 h. The mixture was quenched with 1N HCl to pH 1 and THF was evaporated in vacuo. The suspension was extracted with EtOAc (2×50 mL) and the combined organic extracts dried (Na2SO4). Purification by recrystallization from hexanes-EtOAc (1:1) afforded the title compound (2.4 g) as a white solid. The mother liquor was purified by a Biotage 40S column (eluted 9percent acetone in DCM) to give a second batch of the compound tert-butyl ((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)carbamate (1.1 g). Both batches were combined (total yield 92percent). (0146) 1H NMR: (DMSO-d6) delta ppm 0.96-1.10 (m, 4H), 1.22 (dd, J=5.5, 9.5 Hz, 1H), 1.39 (s, 9H), 1.70 (t, J=5.5 Hz, 1H), 2.19-2.24 (m, 1H), 2.90 (m, 1H), 5.08 (d, J=10 Hz, 1H), 5.23 (d, J=17 Hz, 1H), 5.45 (m, 1H), 6.85, 7.22 (s, NH (rotamers); LC-MS MS m/z 331 (M++H). |
| 92% |
|
A solution of the product of Step 1 (2.62 g, 11.5 mmol) and CDI (2.43 g, 15.0 mmol) in THF (40 mL) was heated at reflux for 50 minutes under nitrogen. The solution was cooled to room temperature and transferred by cannula to a solution of cyclopropylsulfonamide (1.82 g, 15.0 mmol) in THF (10 mL). To the resulting solution was added DBU (2.40 mL, 16.1 mmol) and stirring was continued for 20 hours. The mixture was quenched with 1N HCl to pH 1 and THF was concentrated in vacuo. The suspension was extracted with ethyl acetate (2.x.50 mL) and the combined organic extracts were dried (Na2SO4), filtered, and concentrated. Purification by recystallization from hexanes-ethyl acetate (1:1) afforded the title compound (2.4 g) as a white solid. The mother liquor was purified by a Biotage 40S column (eluted 9percent acetone in dichloromethane) to give a second batch of the title compound (1.1 g). Both batches were combined (total yield 92percent). 1H NMR (DMSO-d6) delta 0.96-1.10 (m, 4H), 1.22 (dd, J=5.5, 9.5 Hz, 1H), 1.39 (s, 9H), 1.70 (t, J=5.5 Hz, 1H), 2.19-2.24 (m, 1H), 2.90 (m, 1H), 5.08 (d, J=10 Hz, 1H), 5.23 (d, J=17 Hz, 1H), 5.45 (m, 1H), 6.85, 7.22 (s, NH (rotamers); MS m/z 331 (M++H). |
| 92% |
|
A solution of the product of Step 1 (2.62 g, 11.5 mmol) and CDI (2.43 g, 15.0 mmol) in THF (40 mL) was heated at reflux for 50 minutes under nitrogen. The solution was cooled to room temperature and transferred by cannula to a solution of cyclopropylsulfonamide (1.82 g, 15.0 mmol) in THF (10 mL). To the resulting solution was added DBU (2.40 mL, 16.1 mmol) and stirring was continued for 20 hours. The mixture was quenched with 1N HCl to pH 1 and THF was concentrated in vacuo. The suspension was extracted with ethyl acetate (2.x.50 mL) and the combined organic extracts were dried (Na2SO4), filtered, and concentrated. Purification by recrystallization from hexanes-ethyl acetate (1:1) afforded the title compound (2.4 g) as a white solid. The mother liquor was purified by a Biotage 40S column (eluted 9percent acetone in dichloromethane) to give a second batch of the title compound (1.1 g). Both batches were combined (total yield 92percent). 1H NMR (DMSO-d6) delta 0.96-1.10 (m, 4H), 1.22 (dd, J=5.5, 9.5 Hz, 1H), 1.39 (s, 9H), 1.70 (t, J=5.5 Hz, 1H), 2.19-2.24 (m, 1H), 2.90 (m, 1H), 5.08 (d, J=10 Hz, 1H), 5.23 (d, J=17 Hz, 1H), 5.45 (m, 1H), 6.85, 7.22 (s, NH (rotamers); MS m/z 331 (M++H). |
| 92% |
|
Step 2:; A solution of the product of Step 1 (2.62 g, 11.5 mmol) and CDI (2.43 g, 15.0 mmol) in THF (40 mL) was heated at reflux for 50 minutes under nitrogen. The solution was cooled to room temperature and transferred by cannula to a solution of cyclopropylsulfonamide (1.82 g, 15.0 mmol) in THF (10 mL). To the resulting solution was added DBU (2.40 mL, 16.1 mmol) and stirring was continued for 20 hours. The mixture was quenched with 1.0M HCl to pH=1, and THF was evaporated in vacuo. The suspension was extracted with ethyl acetate (2.x.50 mL) and the organic extracts were combined and dried (Na2SO4). Filtration, concentration, and purification by recrystallization from hexanes-ethyl acetate (1:1) provideed the desired product (2.4 g) as a white solid. The mother liquor was purified by a Biotage 40S column (eluted 9percent acetone in dichloromethane) to give a second batch of the desired product (1.1 g). Both batches were combined (total yield 92percent). 1H NMR: (DMSO-d6) delta 0.96-1.10 (m, 4H), 1.22 (dd, J=5.5, 9.5 Hz, 1H), 1.39 (s, 9H), 1.70 (t, J=5.5 Hz, 1H), 2.19-2.24 (m, 1H), 2.90 (m, 1H), 5.08 (d, J=10 Hz, 1H), 5.23 (d, J=17 Hz, 1H), 5.45 (m, 1H), 6.85, 7.22 (s, NH (rotamers)); LC-MS, MS m/z 331 (M++H). |
| 85% |
|
A solution of the acid 1d (8.70 g, 38.3 mmol) and carbonyldiimidazole (8.0 g, 50 mmol) in THF (125 mL) was refluxed for 1 h. To a slurry of cyclopropylsulfonamide 1b (6.03 g, 49.8 mmol) in THF (20 mL) was added at room temperature the above solution via cannular, followed by DBU (8.3 mL, 55.5 mmol). The mixture was stirred for 17 h whereupon it was acidified to pH 7 with 1N HCl. The organic solvent was removed in vacuo and ethyl acetate (200 mL) was added. The aqueous layer was further acidified to pH 1, and the organic layer drawn off. The aqueous layer was extracted with ethyl acetate (2.x.150 mL) and the combined organic layers were washed with brine (110 mL), dried over sodium sulfate, and concentrated in vacuo. The crude solid was purified by silica gel chromatography (ethyl acetate/hexanes) to afford 11.0 g of the product 1e in 85percent yield. 1H NMR (300 MHz, CD3OD): delta 5.64-5.52 (m, 1H), 5.29 (d, J=17.1 Hz, 1H), 5.12 (d, J=10.5 Hz, 1H), 2.95 (bs, 1H), 2.29-2.15 (m, 1H), 1.88-1.82 (m, 1H), 1.47 (s, 9H), 1.33-1.00 (m, 5H). |
| 82% |
|
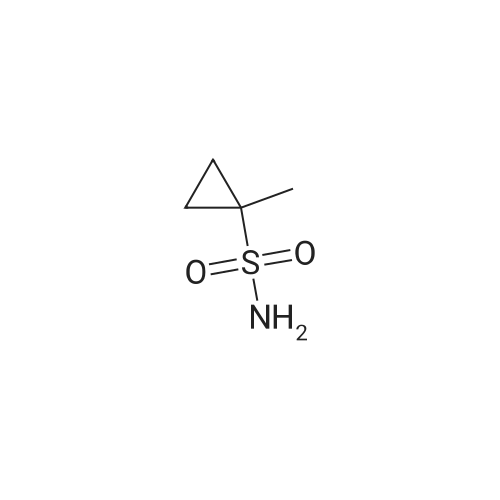
Step B; To a solution of acid 4B2 (567 mg, 2.49 mmol), in THF (20 mL), was added CDI (515 mg, 3.17 mmol). The resulting solution was stirred for 30 min, refluxed for 30 min and allowed to cool down to RT. Cyclopropylsulfonamide 4A4 (455 mg, 3.76 mmol) was added followed by the addition of DBU (0.75 mL, 5.02 mmol) and the reaction was stirred 12 h. The THF was removed in vacuo and the residue was diluted with EtOAc, washed with 1 M HCI (2 X 100 mL) and brine, dried (MgSO4) and purified by flash chromatography (elution conditions: 70:30 hexane/EtOAc) to afford 682 mg (82percent) of compound 4B3 as a white solid. |
| 66% |
|
A solution of compound I-1 (0.52 g, 2.3 mmol), 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluoro-phosphate methanaminium (HATU, 1.74 g, 4.6 mmol), and 4-dimethylaminopyridine (1.39 g, 11.6 mmol) in CH2Cl2 (40 mL) was stirred at room temperature for 1 hour, followed by slow addition of cyclopropanesulfonamide (0.57 g, 4.7 mmol), diisopropylethylamine (1.81 mL, 14.0 mmol), and 1,8-diazabicyclo[5,4,0]undec-7-ene (1.80 g, 11.7 mmol) over 15 minutes. After the reaction mixture was stirred at room temperature overnight, the solvent was removed under vacuum. The residue was purified by silica gel column chromatography to give compound I-2 (0.51 g, 66percent). MS m/z 353.1 (M|+23); 1H NMR (CDCl3) delta 9.75 (brs, 1H), 5.64-5.51 (m, 1H), 5.30 (d, J=17.4 H), 5.16 (d, J=10.2 Hz, 1H), 2.95-2.89 (m, 1H), 2.19-2.10 (m, 1H), 1.93-1.88 (m, 1H), 1.47 (s, 9H), 1.46-1.38 (m, 1H), 1.32-1.23 (m, 2H), 1.15-1.00 (m, 2H). |
| 66% |
|
Example 1Synthesis of {4-Cyclopropanesulfonylaminocarbonyl-2,15-dioxo-18-[2-(4-trifluoromethyl-phenyl)-benzo[4,5]furo[3,2-d]pyrimidin-4-yloxy]-3,16-diaza-tricyclo[14.3.0.04,6]nonadec-14-yl}-carbamic acid cyclopentyl ester (Compound 1)Compound I-3 was first prepared from commercially available 1-t-butoxycarbonylamino-2-vinyl-cyclopropanecarboxylic acid ethyl ester via the route shown below:To a solution of 1-t-butoxycarbonylamino-2-vinyl-cyclopropanecarboxylic acid ethyl ester (0.34 g, 1.3 mmol) in THF (5 mL) and methanol (5 mL) was added a suspension of LiOH (0.13 g, 5.3 mmol) in water (1.4 mL). After being stirred overnight at room temperature, the reaction was quenched with 10percent HCl (2 mL) and the solvent was removed under vacuum. The resultant solid powder was washed with water (10 mL) to give compound I-1 (0.27 g, 90percent). MS m/z 249.9 (M++23); 1H NMR (CDCl3) delta 10.35 (brs, 1H), 5.84-5.71 (m, 1H), 5.29 (d, J=17.4 Hz, 1H), 5.12 (d, J=10.2 Hz, 1H), 2.23-2.14 (m, 1H), 1.87-1.65 (m, 1H), 1.58-1.41 (m, 1H), 1.43 (s, 9H).A solution of compound I-1 (0.52 g, 2.3 mmol), 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluoro-phosphate methanaminium (HATU, 1.74 g, 4.6 mmol), and 4-dimethylaminopyridine (1.39 g, 11.6 mmol) in CH2Cl2 (40 mL) was stirred at room temperature for 1 hour, followed by slow addition of cyclopropanesulfonamide (0.57 g, 4.7 mmol), diisopropylethylamine (1.81 mL, 14.0 mmol), and 1,8-diazabicyclo[5,4,0]undec-7-ene (1.80 g, 11.7 mmol) over 15 minutes. After the reaction mixture was stirred at room temperature overnight, the solvent was removed under vacuum. The residue was purified by silica gel column chromatography to give compound I-2 (0.51 g, 66percent). MS m/z 353.1 (M++423); 1H NMR (CDCl3) delta 9.75 (brs, 1H), 5.64-5.51 (m, 1H), 5.30 (d, J=17.4H), 5.16 (d, J=10.2 Hz, 1H), 2.95-2.89 (m, 1H), 2.19-2.10 (m, 1H), 1.93-1.88 (m, 1H), 1.47 (s, 9H), 1.46-1.38 (m, 1H), 1.32-1.23 (m, 2H), 1.15-1.00 (m, 2H).To a solution of compound I-2 (0.50 g, 1.5 mmol) in MeOH (8 mL) was added SOCl2 (0.26 g, 2.2 mmol) at room temperature. After the reaction mixture was refluxed for 1 hour, MeOH and SOCl2 was removed under vacuum. The residue was triturated from pentane and filtered to give intermediate I-3 as an off-white solid (0.32 g, 91percent). MS m/z (M++1); 1H NMR (CD3COD) delta 5.77-5.65 (m, 1H), 5.43 (d, J=17.4 Hz, 1H), 5.32 (d, J=10.2 Hz, 1H), 3.06-2.97 (m, 1H), 2.45 (dd, J=17.4 Hz, J=7.8, 1H), 2.16 (dd, J=8.0 Hz, J=7.8 Hz, 1H), 1.75 (dd, J=10.1 Hz, J=7.8 Hz, 1H), 1.32-0.86 (m, 4H).Compound 1 was prepared via the route shown below:A solution of 3-amino-benzofuran-2-carboxylic acid amide (1.00 g, 5.7 mmol) and pyridine (1 mL, 12.26 mmol) in THF (25 mL) was stirred at 0° C. for 10 min. To the resulting solution was slowly added 4-trifluoromethyl-benzoyl chloride (1.48 g, 7.1 mmol). Then the temperature was raised to room temperature and the mixture was stirred for 12 h. After the solvent was removed under reduced pressure, the resulting solid was collected, washed with water, and air-dried to yield I-4 (1.92 g, 96.0percent). MS: m/z 349.0 (M++1).To a suspension of I-4 (1.92 g, 5.5 mmol) and 2N NaOH (13 mL) in EtOH (25 mL) was heated at 85° C. for 12 h. After cooled, the mixture was acidified and then EtOH was removed. The resulting solid was collected, filtrated, washed with water, and dried to afford I-5 (1.71 g, 95.0percent). MS m/z 331 (M++1).A solution of I-5 (1.71 g, 5.2 mmol) and excess phosphorus oxychloride (POCl3) was refluxed for 2 hours. After cooled and thoroughly concentrated, the mixture was subjected to extraction with methylene chloride and 10percent sodium hydroxide. The organic layer was dried over MgSO4, concentrated, and crystallized from CH2Cl2 and n-hexane to give compound I-6 (1.49 g, 82percent). MS m/z 348.8, 350.9 (M++1); 1H NMR (CDCl3) delta 8.70 (d, 2H), 8.34 (d, 1H), 7.82-7.75 (m, 4H), 7.57 (ddd, 1H).To a suspension of boc-trans-4-hydroxy-L-proline (0.53 g, 2.3 mmol) in DMSO (25 mL) was added t-BuOK (0.82 g, 5.1 mmol) at 0° C. After the mixture was allowed to warm to room temperature and stirred for 1 hour, compound I-6 (0.81 g, 2.3 mmol) was added slowly at 10° C. Stirring was continued overnight. Iodomethane (1.02 g, 6.9 mmol) was added and the reaction mixture was stirred at room temperature for additional 30 minutes. The reaction mixture was neutralized to pH 6-7 by 10percent HCl aqueous solution and subjected to extraction with methylene chloride. The organic layer was dried over MgSO4, evaporated under vacuum, and purified by silica gel column chromatography to give compound I-7 (1.12 g, 86percent). MS m/z 557.8 (M++1); 1H NMR (CDCl3) delta 8.63 (d, 2H), 8.28 (d, 1H), 7.80-7.74 (m, 2H), 7.70 (d, 2H), 7.51 (ddd, 1H).To a solution of compound I-7 (1.13 g, 2.0 mmol) in MeOH (20 mL) was added SOCl2 (1.21 g, 9.8 mmol) at room temperature. The reaction mixture was refluxed for 1 hour, and MeOH and SOCl2 were removed. The residue was triturated in penta... |
| 66% |
|
A solution of compound 1-1 (0.52 g, 2.3 mmol), 2-(lH-7-azabenzotriazol-l-yl)- 1,1 ,3,3-tetramethyl uronium hexafluoro -phosphate methanaminium (HATU, 1.74 g, 4.6 mmol), and 4-dimethylaminopyridine (1.39 g, 1 1.6 mmol) in CH2CI2 (40 mL) was stirred at room temperature for 1 hour, followed by slow addition ofcyclopropanesulfonamide (0.57 g, 4.7 mmol), diisopropylethylamine (1.81 mL, 14.0 mmol), and l ,8-diazabicyclo[5,4,0]undec-7-ene (1.80 g, 11.7 mmol) over15 minutes. After the reaction mixture was stirred at room temperature overnight, the solvent was removed under vacuum. The residue was purified by silica gel column chromatography to give compound 1-2 (0.51 g, 66percent). MS m/z 353.1 (M++23); !H NMR (CDCI3) d 9.75 (brs, 1H), 5.64-5.51 (m, 1H), 5.30 (d, J = 17.4 H), 5.16 (d, J = 10.2 Hz, 1H), 2.95-2.89 (m, 1H), 2.19-2.10 (m, 1H), 1.93-1.88 (m, 1H), 1.47 (s, 9H), 1.46-1.38 (m, 1H), 1.32-1.23 (m, 2H), 1.15-1.00 (m, 2H). |
|
|
To a solution of Boc-D-beta-vinyl cyclopropane amino acid (4.5g) in DMF was added CDI (3.97g). The reaction mixture was stirred at 4O0C for Ih and then added cyclopropylsulfonamide (4.66g) and DBU (5.78ml). The reaction mixture was stirred overnight at 4O0C. The reaction mixture was extracted with EtOAc. The organic extracts were washed with IM NaHCO3, brine, dried over Na2SO4, filtered and concentrated. The residue was desolved in 4NHCL/Dioxane. The reaction was stirred at RT for lhour and then concentrated in vacuo. The resulted solid was carrired out to next step without further purification. MS (ESI): m/z = 231.10 [M+H]. |
|
With hydrogenchloride; 1,8-diazabicyclo[5.4.0]undec-7-ene; 1,1'-carbonyldiimidazole; In tetrahydrofuran; [(2)H6]acetone; |
Step 1: tert-butyl ((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)carbamate A solution of <strong>[159622-10-3](1R,2S)-1-((tert-butoxycarbonyl)amino)-2-vinylcyclopropanecarboxylic acid</strong> (2.62 g, 11.5 mmol) and CDI (2.43 g, 15.0 mmol) in THF (40 mL) was heated at reflux for 50 min under nitrogen. The solution was cooled to room temperature and transferred by cannula to a solution of cyclopropylsulfonamide (1.82 g, 15.0 mmol) in THF (10 mL). To the resulting solution was added DBU (2.40 mL, 16.1 mmol) and stirring was continued for 20 h. The mixture was quenched with 1N HCl to pH 1 and THF was evaporated in vacuo. The suspension was extracted with EtOAc (2*50 mL) and the combined organic extracts dried (Na2SO4). Purification by recystallization from hexanes-EtOAc (1:1) afforded the title compound (2.4 g) as a white solid. The mother liquor was purified by a Biotage 40S column (eluted 9percent acetone in DCM) to give a second batch of the compound tert-butyl ((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)carbamate (1.1 g). |
| 2.8 g |
|
Intermediate C6: tert-butyl (1R,2S)-1-(cyclopropylsulfonylcarbamoyl)-2-vinylcyclopropylcarbamate (0154) (0155) Following the procedure in Scheme III, Intermediate C5 (6.0 g, 23.5 mmol) was added in a solution of LiOH (3.0 g, 73.5 mmol) in THF/MeOH/H2O (20 mL/20 mL/10 mL). After 1 h at r.t., the pH was adjusted to 4 with 1N HCl, aqueous layer was separated, EA was added, and the mixture was extracted with EA twice. The combined organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to give the yellow oil, which was used directly in the next reaction without further purification. (0156) Then the intermediate (5.6 g, 24.2 mmol) and CDI (5.2 g, 31.5 mmol) were dissolved in THF (20 mL). After refluxed for 1 h, the mixture was cooled to r.t. and then a solution of cyclopropanesulfonamide (3.8 g, 31.5 mmol) in DCM (30 mL) was added followed by adding DBU (5.2 mg, 34.0 mmol). The mixture was stirred overnight at the room temperature, concentrated and extracted with EA twice. The combined organic layer was washed with brine, dried over Na2SO4, filtered, concentrated and purified by flash column chromatography to give the title compound C6 (2.8 g, 36.4percent) as white solid. (0157) 1H NMR (400 MHz, CDC3) delta 9.52 (s, 1H), 5.66-5.57 (m, 1H), 5.32 (d, J=13.2 Hz, 1H), 5.17 (dd, J=10.4, 0.8 Hz, 1H), 2.94-2.87 (m, 1H), 2.17 (q, J=8.4 Hz, 1H), 1.92-1.89 (m, 1H), 1.48 (s, 9H), 1.45-1.39 (m, 1H), 1.32-1.25 (m, 2H), 1.13-1.00 (m, 2H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping