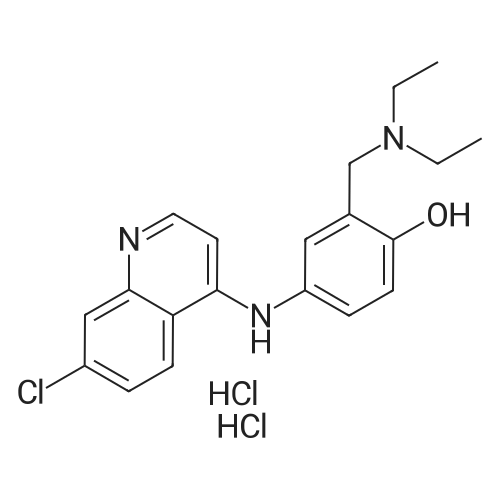
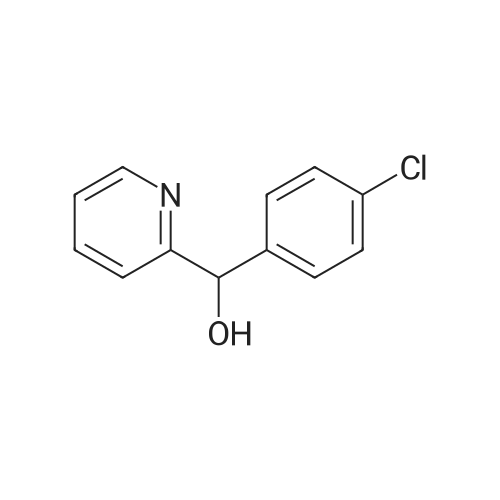
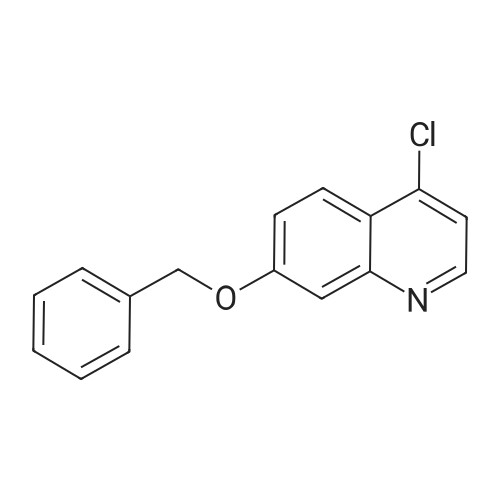
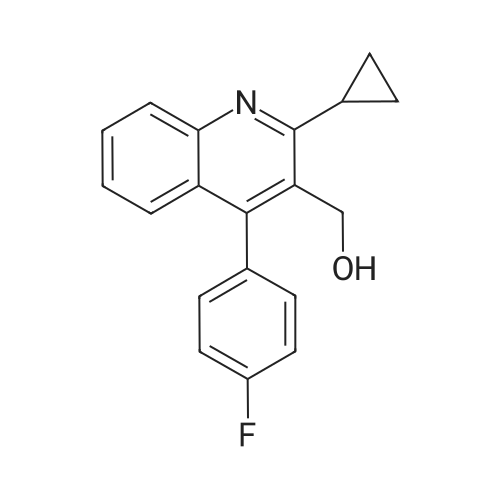
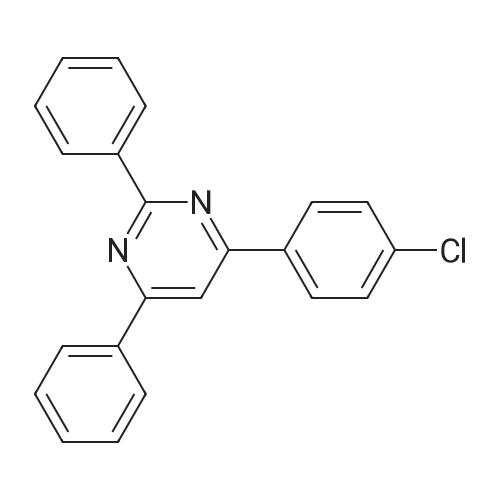
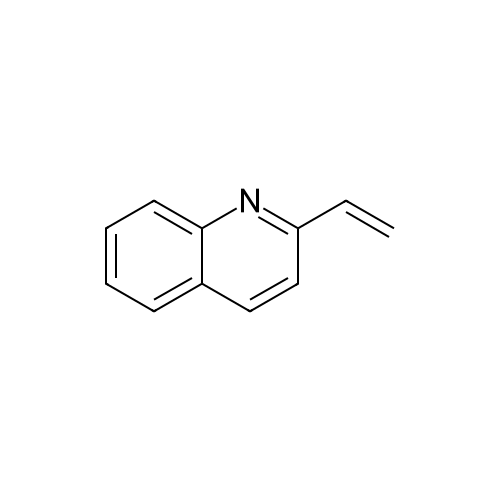
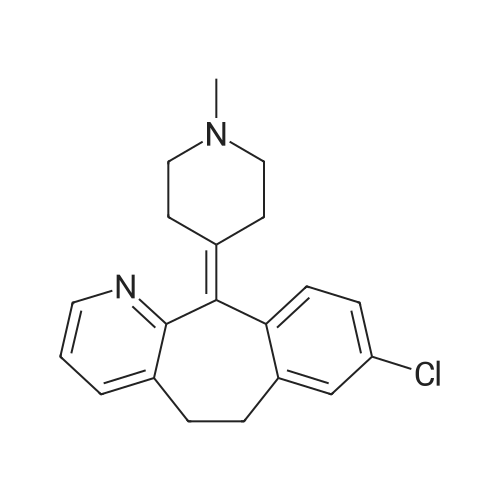
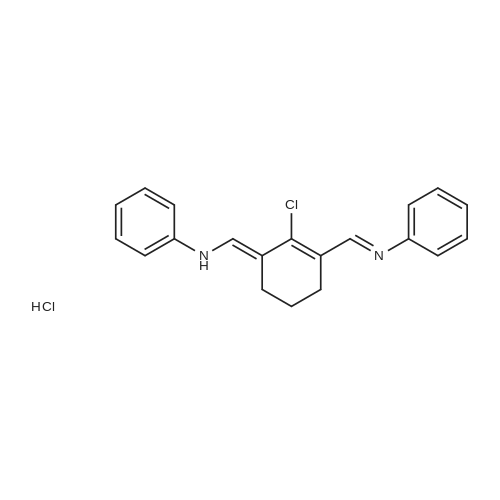
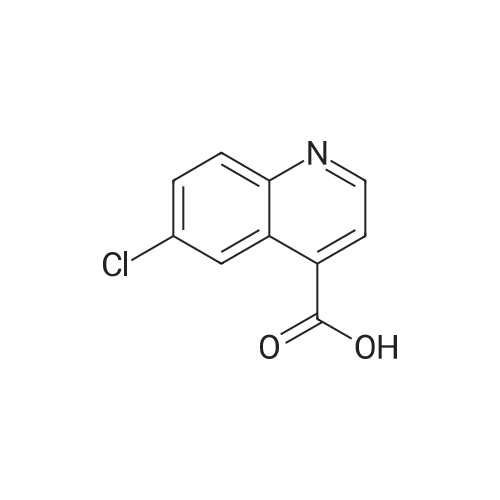
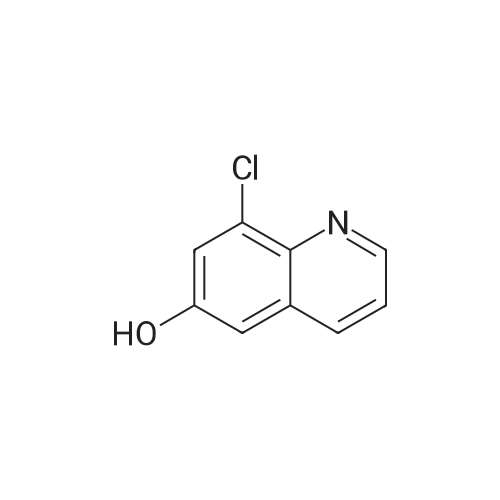
| 89% |
In toluene; at 5 - 25℃; for 2.0h;Heating / reflux;Purification / work up; |
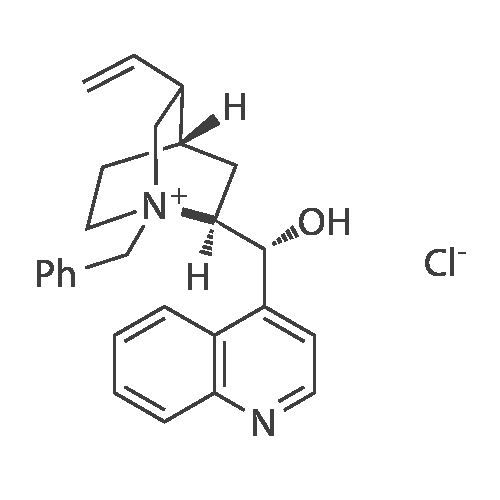
Example 1 - Purification of Optically Impure Compound Till) by Crystallization from Toluene[0029] A reaction vessel equipped with a thermometer, a magnetic stirrer, and a condenser was charged with compound (III) (50 g) which contained 1.1% of the (R)-compound (i.e., 97.8% ee) and toluene (250 mL). The resulting mixture was heated to reflux. After the impure compound (III) was dissolved, the resulting solution was allowed to cool to 250C, then stirred at 25C for 1 hour. Then, the solution was cooled to 5C and stirred at that temperature for 1 hour. A solid precipitated from the solution as crystals, and the resulting crystals were filtered, washed with cold toluene, then dried in vacuum at 40C for 16 hours to afford 44.5 g compound (III) (89% yield) having 99.61% (S) enantiomer and 0.39% (R)-enantiomer (99.2% ee), as measured by HPLC.Example 2 - Purification of Compound (III) by a Second Crystallization from Toluene[0030] A reaction vessel equipped with a thermometer, a magnetic stirrer, and a condenser was charged with compound (III) (44.5 g), resulting from Example 1, above, and toluene (222 mL). The resulting mixture was heated to reflux. After the compound was dissolved, the solution was allowed to cool to 25C, then stirred at 25C for 1 hour. Subsequently, the solution was cooled to 5C, then stirred at 5C for 1 hour. A solid precipitated from solution as crystals, and the resulting crystals were filtered, washed with cold toluene, and dried in vacuum at 40C for 16 hours to afford 39.6 g compound (III) (89% yield) having 99.82% (S)- enantiomer and 0.18% (R)-enantiomer (99.6% ee), as measured by HPLC. |
|
|
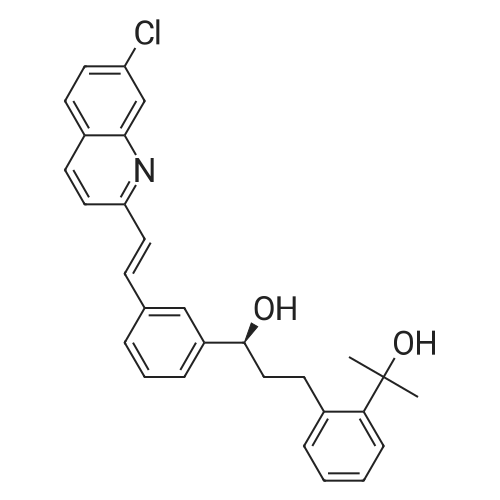
Step 3 2-(2-(3(S)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-hydroxypropyl)phenyl)-2-propanol To a solution of the hydroxyester of Step 2 (38.68 g, 84.36 mmol) in 600 mL of toluene at 0 C. was added slowly 225 mL of 1.5M MeMgBr in toluene:THF 3:1 and the mixture was stirred at r.t. for 4 h. It was then poured into 2 L cold 12% NH4 OAc and 25 mL of AcOH were added. The products were extracted in EtOAc, washed with brine, dried over Na2 SO4 and concentrated. Flash chromatography of the residue with EtOAc:toluene 15:85 and 25:75 afforded first the methyl ketone derivative, then the title compound. Yield 24.06 g, 62%. 1 H NMR (CD3 COCD3) delta 1.59 (3H, s), 1.62 (3H, s), 2.11 (2H, m), 3.16 (2H, td), 4.15 (1H, s, OH), 4.52 (1H, d, OH), 4.81 (1H, m), 7.04-7.28 (3H, m), 7.37-7.57 (5H, m), 7.60 (1H, m), 7.78 (1H, s), 7.83-8.02 (4H, m), 8.32 (1H, d). |
|
|
Step 3 2-(2-(3(S)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-hydroxypropyl)phenyl)-2-propanol To a solution of the hydroxyester of Step 2 (38.68 g, 84.36 mmol) in 600 mL of toluene at 0 C. was added slowly 225 mL of 1.5M MeMgBr in toluene:THF 3:1 and the mixture was stirred at r.t. for 4 h. It was then poured into 2 L cold 12% NH4 OAc and 25 mL of AcOH were added. The products were extracted in EtOAc, washed with brine, dried over Na2 SO4 and concentrated. Flash chromatography of the residue with EtOAc:toluene 15:85 and 25:75 afforded first the methyl ketone derivative, then the title compound. Yield 24.06 g, 62%. 1 H NMR (CD3 COCD3) delta 1.59 (3H, s), 1.62 (3H, s), 2.11 (2H, m), 3.16 (2H, td), 4.15 (1H, s, OH), 4.52 (1H, d, OH), 4.81 (1H, m), 7.04-7.28 (3H, m), 7.37-7.57 (5H, m), 7.60 (1H, m), 7.78 (1H, s), 7.83-8.02 (4H, m), 8.32 (1H, d). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping