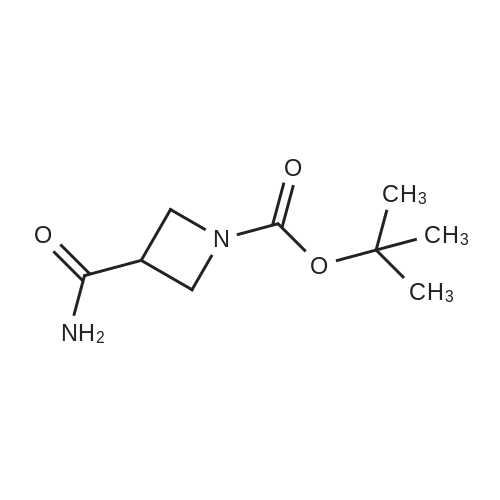
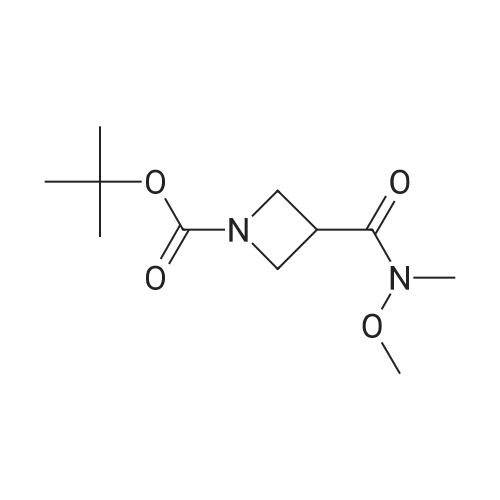
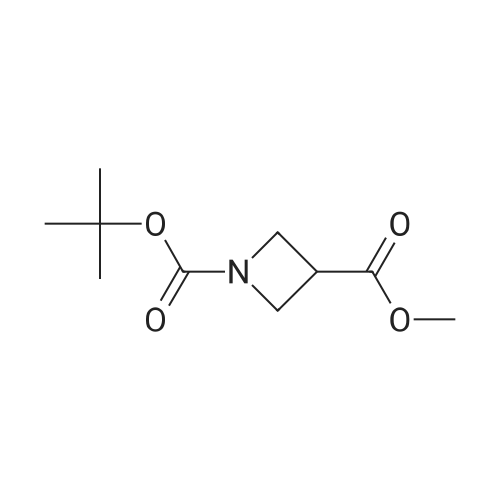
| 99% |
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; for 20h; |
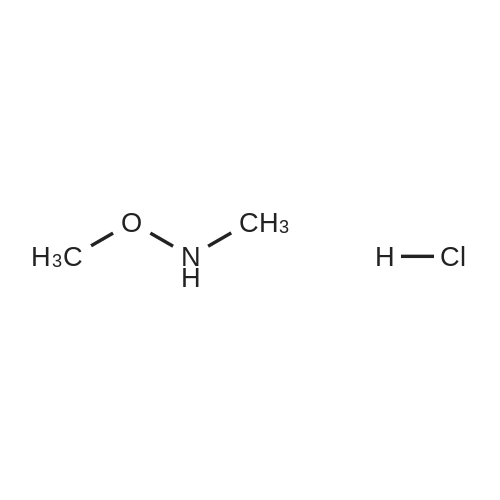
DMF (40 mL) was added N,O-dimethylhydroxylamine hydrochloride (2.89 g, 29.8 mmol), EDC (5.55 g, 29.8 mmol), HOBt (3.36 g, 24.9 mmol) and DIPEA (9.82 mL, 54.7 mmol). The reaction was stirred at room temperature for 20 h. The reaction was diluted with water (150 mL) and extracted with EtOAc (2 x 100 mL). The combined organic extracts were washed with water (150 mL) and brine (150 mL), dried over MgSO4 and concentrated under vacuum to afford 4 as a clear colorless oil (6.06 g, 99%); Η-nmr (400 MHz, CDCI3) δ 4.21-4.11 (m, 2H), 4.05 (t, J= 8.0 Hz, 2H), 3.66 (s, 3H), 3.68-3.59 (m, IH), 3.21 (s, 3H), 1.44 (s, 9H); m/z 189.1 [M-1Bu]. |
| 99% |
|
To a solution of 1-(te/-bυtoxycarbonyl)azetidine-3-carboxylic acid (2.00 g, 9.94 mmol) in THF (50 mL) was added EDCI (2.10 g, 10.93 mmol), HOBt (1.48 g, 10.93 mmol), and /V,/V-diisopropylethylamine (5.19 mL, 29.82 mmol). The mixture was stirred at rt for 15 min. Λ/,Odimethylhydroxylamine hydrochloride (1.16 g, 11.93 mmol) was added and stirring continued for 64 h. The crude reaction was purified via ISCO chromatography using 3:1 ethyl/hexanes to afford 2.4 g (99%) of the desired product, which was used without further characterization. |
| 99% |
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 16h;Inert atmosphere; |
1-Boc-azetidine-3-carboxylic acid (5 g; 24.9 mmol) and N,O-dimethylhydroxylaminehydrochloride (3.64 g; 37.3 mmol) were placed in a round bottom flask under N2. DCM(75 mL) was added, followed by EDCI.HC1 (7.15 g; 37.3 mmol), DMAP (155 mg; 1.27mmol) and DIPEA (6.5 mL, 37.4 mmol). The reaction mixture was stirred at RT for 16h and diluted with DCM (100 mL). The organic layer was washed with aqueous 1M HC1 (2 x 50 mL), sat. NaHCO3 solution (50 mL), and brine (50 mL). The organic phasewas decanted, dried over MgSO4, filtered, and evaporated to dryness yielding 6.04 g(99%) of intermediate 73. |
| 98% |
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; |
A mixture of 298 (1.8 g, 9 mmol), N,O-dimethylhydroxylamine hydrochloride (2.6 g, 27 mmol), EDCI (5 g, 27 mmol), HOBt (0.1 g, 1 mmol), and DIPEA (12.5 mL, 72 mmol) in DMF (30 mL) was stirred at 20C overnight. The reaction was then concentrated to half volume in vacuo, poured onto water, and extracted three times with ethyl acetate. The combined organic phases were washed with saturated aqueous NH4Cl, saturated aqueous NaHCO3, water and brine, dried (Na2SO4), and concentrated to give 299 as a clear oil (2.1 g, 98%). |
| 84% |
With 1,1'-carbonyldiimidazole; In dichloromethane; at 20℃; for 17h; |
A solution of 1-boc-azetidine-3-carboxylic acid (5.00 g, 24.8 mmol, and CDI (4.23 g, 26.1 mmol) in dichloromethane (100 mL) was stirred at room temperature for 1 hour, then N,O-dimethylhydroxylamine hydrochloride (4.0 g, 29.8 mmol) was added and stirring continued at room temperature for 16 hours. The resulting suspension was washed with water (3×30 mL), sat. aq. NaHCO3 (3×30 mL), and brine (3×30 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated to give tert-butyl 3-(methoxy(methyl)carbamoyl)azetidine-1-carboxylate (87a, 5.1 g, 84% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 4.14 (br s, 2H), 4.05 (t, J=8.6 Hz, 2H), 3.66 (s, 3H), 3.65 (m, 1H), 3.20 (s, 3H), 1.43 (s, 9H). |
| 83% |
With triethylamine; HATU; In dichloromethane; N,N-dimethyl-formamide; at 20℃; for 16h; |
Example 232: 3-[5-Bromo-2-(5-trifluoromethyl-furan-2-ylmethoxy)-benzv?- azetidine.; Step A: Preparation of 3-(Methoxy-methyl-carbamoyl)-azetidine-1 - carboxylic acid tert-butyl ester.; To a solution of azetidine-1 ,3-dicarboxylic acid mono-tert-butyl ester (3.5 g, 17 mmol) in DMF (50 ml_) was added O1N- dimethyl-hydroxylamine hydrochloride (3.4 g, 34 mmol), thethylamine (9.6 mL, 69 mmol), HATU (13.4 g, 34.6 mmol) and DCM (125 mL). After stirring for 16 h, saturated NaHCO3 solution and ethyl acetate were added. The aqueous portion was extracted three times with ethyl acetate. The combined organic fractions were dried (Na2SO4) and concentrated and the crude product was purified using RP HPLC (basic conditions) to provide the title compound (3.5 g, 83%) MS (ESI): mass calcd. for CnH2ON2O4, 244.1 ; m/z found, 189.1 [M-t- Bu]+. 1H NMR (CDCI3): 4.14-4.03 (m, 2H), 4.05 (t, J = 8.7 Hz, 2H), 3.66 (s, 3H), 3.63-3.59 (m, 1 H), 3.21 (s, 1 H), 1.43 (s, 9H). |
| 80% |
With 4-methyl-morpholine; O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate; In N,N-dimethyl-formamide; at 20℃; for 16h; |
To a stirred solution of l-(tert-butoxycarbonyl)azetidine-3-carboxylic acid (3.0 g, 14.9 mmol, CAS 142253-55-2) in DMF (45 mL) at room temperature was added N,0-dimethyl hydroxylamine hydrochloride (1.75 g, 17.9 mmol), HBTU (8.48 g, 22.4 mmol) and iV-methyl morpholine (6.56 mL, 59.6 mmol). After 16 hours, the reaction mixture was poured into a 1: 1 mixture of water and saturated NH4C1 and extracted with EtOAc. The combined organic extracts were washed with saturated aqueous NaHC03 and brine, dried (Na2S04) and concentrated in vacuo to afford the title compound (2.9 g, 80%) as a colorless oil. MS (ISP): 189.1 ([M- C4H8+H]+). |
| 77% |
With bis-(2-oxo-3-oxazolidinyl)phosphoryl chloride; N-ethyl-N,N-diisopropylamine; In DMF (N,N-dimethyl-formamide); at 20℃; for 2h; |
Intermediate 24: tert-Butvl 3- {r methoxy( methyl) amino 1 carbonyl } azetidine-l-carboxylate; To a solution of 1-(tert-butoxycarbonyl)azetidine-3-carboxylic acid (H. Itani et al, Biorg. Med. Chem. Lett. 12 (5), 757-762,2002) (0.5 g, 2.48 mmol) and bis (2-oxo-3- oxazolidinyl) phosphinic chloride (0.633 g, 2.48 mmol) in DMF (4 mL) was added N,O- dimethylhydroxylamine hydrochloride (339 mg, 3.48 mmol), followed by diisopropylethyl amine (1.3 mL, 7.45 mmol). The exothermic reaction was allowed to cool to room temperature and was stirred for 2 hours. Excess base was removed under reduced pressure, the residue was diluted with ethyl acetate (100 mL), washed with potassium phosphate buffer (1M, pH 7,2x 100 mL) and with water (2x 100 mL) and dried over sodium sulfate. Chromatography on silica gel with hexanes/ acetone (3:1) gave 470 mg (77%) of the product as a colourless solid. MS (ESP): 267.23 (MNa+) for C11H20N2O4 'H-NMR (DMSO-d(at) 5: 1.36 (s, 9H); 3.10 (s, 3H); 3.61 (s, 3H); 3.68 (m, 1H); 3.83-4.00 (m, 4H). |
| 76% |
With triethylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate; In dichloromethane; for 16h; |
A solution of R-1 (50 g, 250 mmol) and HATU (104 g, 270 mmol) in CH2C12 (8000 mL) is treated with TEA (135 mL, 1000 mmol). The mixture is stirred for 16 h then washed with saturated aqueous ammonium chloride and filtered through a phase separator. The organics are collected and volatiles are removed in vacuo to afford a crude residue that is purified by flans chromatography (Si02, 12% EtOAc in heptane to 100%EtOAc) to afford 1-1 (46 g, 76%) m/z 245.1 [M+H]. |
| 72% |
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; triethylamine; In N,N-dimethyl-formamide; at 25℃; for 16h;Inert atmosphere; |
A mixture of l-Boc-azetidine-3-carboxylic acid (50 g, 248 mmol), triethylamine (69.3 mL, 497 mmol), 1 -hydroxybenzotriazole (33.5 g, 248 mmol) and EDC1 (47.6 g, 248 mmol) and (9, /V-dimcthyl hydroxy laminc HC1 (24.24 g, 248.5 mmol) in DMF (1000 mL) was stirred at 25 C for 16 h. The mixture was concentrated in vacuo to give a residue, which was neutralized by HC1 (1M) to pH = 7 and extracted with ethyl acetate (3 x 200 mL). The combined organic layers were washed with NaHCO, (2 x sat.aq. 200 mL), dried over Na2S04 and concentrated in vacuo to give tert- butyl 3- (1025) [methoxy(methyl)carbamoyl]azetidine-l-carboxylate (55 g, 72%) as colorless oil; LC-MS: 189.1 [M-56+H]+. |
|
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine; In dichloromethane; at 0 - 20℃; for 6h; |
To a solution of 980 mg (4.87 mmol) of boc-azetidine-3-carboxylic acid, 951 mg (9.75 mmol) OF N, O- dimethylhydroxylamine hydrochloride, 329 mg (2.435 mmol) of HOBT and 1.87g (9.75 mmol) of 1- [3- (dimethylamino) propyl] -3-ethylcarbodiimide hydrochloride in 30 mL of CH2CL2 was added 2.54 mL (44.61 mmol) of N, N-diisopropylethylamine at 0 oC and it was stirred for 6h at rt. Then the reaction mixture was poured into 100 mL of ether and washed with 20 mL of aq NAHC03. The organic layer was dried over Na2S04 and concentrated. to afford the title compound. 1NMR (CDC13) 8 1.46 (s, 9H), 3.23 (s, 3H), 3.62 (m, 1H), 3.68 (s, 3H), 4.07 (M, 2H), 4.09 (m, 2H). |
|
|
EXAMPLE 6; 1 - [5 -fluoro-2-(methylsulfonyl)benzoyl] -3 -( 1 -methyl- 1 - { [3 - (trifluoromethyl)phenyl] sulfonyl } ethyl)azetidineStep 1 : tert-butyl 3-[methoxy(methyl)amino]carbonyl}azetidine-l-carboxylateTo a 100 ml round bottom flask was added l-(fert-butoxycarbonyl)azetidine-3- carboxylic acid (3.75 g, 18.6 mmol) and 20 ml tetrahydrofuran, followed by CDI (3.63 g, 22.4 mmol). Vigorous gas evolution was observed. After gas evolution stopped, the reaction mixture was stirred at room temperature for additional 30 minutes. Methoxy(methyl)ammonium chloride (2.55 g, 26.1 mmol) was added, followed by diisopropylethyl amine (6.5 ml, 37.3 mmol). The resulting reaction mixture was stirred at room temperature overnight. It was diluted with 120 ml ether and washed with 60 ml saturated NH4Cl. The organics were dried over sodium sulfate, filtered and concentrated. The residue was purified on silica gel column, eluted with 1 :2 to 3:2 ethyl acetate/hexane to give 3.6 g desired product was colorless oil. <n="54"/>1H-NMR (CDCl3): 54.17 (m, 2H), 4.08 (t, J = 8.7 Hz, 2H), 3.69 (s, 3H), 3.5 (b, IH), 3.24 (s, 3H), 1.47 (s, 9H). |
|
With triethylamine; HATU; at 20℃; for 19h; |
EXAMPLE 8; F5-(2- 13 - [2-(Trifluoromethyl)benzovnazetidin- 1 -vU - 1 ,3 -thiazol-5 -yl)-2//-tetrazol-2-yllacetic acid; Step 1 : ferf -Butyl 3 - { [methoxy (methyl)amino] carbonyl ) azetidine- 1 -carboxylate; To a solution of l-(/er/-butoxycarbonyl)azetidine-3-carboxylic acid (3.78 g, 18.8 mmol), N, 0-dimethylhydroxylamine hydrochloride (2.75 g, 28.2 mmol), and Et3N (7.85 mL, 56.4 mmol) was added HATU (7.86 g, 20.7 mmol). The resulting mixture was stirred at room temperature for 19 h. A second portion of HATU (4.5 g, 11.8 mmol) was added and the reaction was stirred at room temperature for 19 h. The mixture was poured into a 250 mL separatory funnel containing water (150 mL) and extracted with EtOAc (2 x 50 mL). The combined organic layers were washed with water, brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography through silica gel, eluting with 20% EtOAc in hexanes to 70% EtOAc in hexanes as a gradient, to afford the title compound as a colorless oil. |
|
With triethylamine; HATU; at 20℃; for 38h; |
Step 1: tert-Butyl 3-[methoxy(methyl)amino]carbonyl}azetidine-1-carboxylate To a solution of 1-(tert-butoxycarbonyl)azetidine-3-carboxylic acid (3.78 g, 18.8 mmol), N,O-dimethylhydroxylamine hydrochloride (2.75 g, 28.2 mmol), and Et3N (7.85 mL, 56.4 mmol) was added HATU (7.86 g, 20.7 mmol). The resulting mixture was stirred at room temperature for 19 h. A second portion of HATU (4.5 g, 11.8 mmol) was added and the reaction was stirred at room temperature for 19 h. The mixture was poured into a 250 mL separatory funnel containing water (150 mL) and extracted with EtOAc (2*50 mL). The combined organic layers were washed with water, brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography through silica gel, eluting with 20% EtOAc in hexanes to 70% EtOAc in hexanes as a gradient, to afford the title compound as a colorless oil. |
|
With triethylamine; dicyclohexyl-carbodiimide; In tetrahydrofuran; at 20℃; for 16h; |
[0299] To a solution of 1-(tert-butoxycarbonyl)azetidine-3-carboxylic acid (5.15 g, 25.6 mmol)in THF (100 mL) was added DCC (7.11 g, 34.5 mmol), Et3N (5.18 g, 51.2 mmol) andN,Odimethylhydroxylaminehydrochloride (3.44 g, 35.3 mmol), the reaction was stirred at RT forabout 16 hr. Concentrated under reduced pressure to remove solvent, the residue was portionedbetween EA (100 mL) and water (50 mL), the aqueous was further extracted with EA (50 mL x3). The combined organic phases were washed with brine (20 mL), concentrated under reducedpressure to remove solvent, then purified by column chromatography on silica gel (200-300mesh, CH2Cb/MeOH = 2011) to give the crude product ( -8.0 g) as a colorless oil. MS (ESI) m/e[M+23t 266.9, [M-55t 189.0. |
| 7.30 g |
|
(1) Synthesis oftert-butyl 3-[methoxy(methyl)carbamoyl]azetidine-1-carboxylate To a solution of 1-(tert-butoxycarbonyl)azetidine-3-carboxylic acid (5.00 g) in tetrahydrofuran (62.1 mL), 1,1'-carbonyldiimidazole (6.05 g) was added and the mixture was stirred at room temperature for an hour. To the reaction mixture, a solution of N,O-dimethylhydroxylamine hydrochloride (3.64 g) and triethylamine (4.02 g) in acetonitrile (62.1 mL) was added and the mixture was stirred at the same temperature for 15 hours. The reaction mixture was concentrated under reduced pressure and water was added to the resulting residue. Extraction was conducted with ethyl acetate and the combined organic layers were washed with an aqueous solution of 5% citric acid and saturated brine. The washed organic layers were dried over anhydrous sodium sulfate and after removing the desiccant by filtration, the filtrate was concentrated under reduced pressure to give tert-butyl 3-[methoxy(methyl)carbamoyl]azetidine-1-carboxylate as a pale yellow oil (7.30 g). 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.43 (s, 9 H) 3.21 (s, 3 H) 3.56 - 3.68 (m, 4 H) 4.00 - 4.09 (m, 2 H) 4.09 - 4.19 (m, 2 H). |
|
With triethylamine; dicyclohexyl-carbodiimide; In tetrahydrofuran; at 20℃; for 16h; |
To a solution of 1-(tert-butoxycarbonyl)azetidine-3-carboxylic acid (5.15 g, 25.6 mmol) in THF (100 mL),Add DCC (7.11g, 34.5 mmol),Et3N (5.18 g, 51.2 mmol)And N,O-dimethylhydroxylamine hydrochloride (3.44 g, 35.3 mmol),The reaction was stirred at room temperature for about 16 hours.Concentration under reduced pressure to remove the solvent, the residue was partitioned between EA (100 mL) and water (50 mL) and the aqueous phase was further extracted with EA (50 mL x 3). The organic phases were combined, washed with saturated brine (20 mL), concentrated under reduced pressure to remove the solvent, and then purified on a tannin gel column (200-300 mesh CH 2 Cl 2 /MeOH=20/1).The crude product was obtained as a colorless oil (-8.0 g). |
|
With (benzotriazo-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 0 - 20℃; for 18h; |
Example 5 1) Synthesis of N-t.butoxycarbonyl-3-(methoxy-methyl-carbamoyl)-azetidine 2 g (10 mmol) of N-t.butoxycarbonyl-azetidine-3-carboxylic acid is dissolved in 20 ml of dimethylformamide under argon. This solution is cooled down to 0 C. using an ice bath and 5.46 g (12.3 mmol) of benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate, 1.4 g (14.3 mmol) of N,O-dimethylhydroxylamine chloride and 6 ml of diisopropylethylamine are added. The reaction mixture is left to return to ambient temperature and is stirred for 18 hours at ambient temperature. The dimethylformamide and diisopropylethylamine are evaporated off under reduced pressure (2 kPa). Purification is carried out by chromatography on silica gel eluding with a heptane/ethyl acetate 50:50 mixture 2.1 g of colourless oil is recovered. TLC: Rf=0.25 (silica gel, eluent: heptane/ethyl acetate 50:50 1H-NMR (CDCl3): δ 1.44 (s, 9H, tBu); 3.22 (s, 3H, -N-C); 3.64 (m, 1H, H3); 3.67 (s, 3H, -O-C); 4.05 and 4.15 (m, 4H, H2 and H2') |
|
|
Carbonyldiimidazole (24.4 g, 150 mmol) was added in portions to a solution of 1-tert-butoxycarbonylazetidine-3-carboxylic acid (23.2 g, 115 mmol) in THF (250 ml) and the mixture was stirred at room temperature for 1.5 h. A suspension of N,O-dimethylhydroxylamine hydrochloride (15.0 g, 154 mmol) in a mixture of acetonitrile (300 ml) and triethylamine (22.0 ml, 162 mmol) was added and the reaction was stirred at room temperature for 24 h. The solvents were evaporated and the residue was partitioned between water (300 ml) and ethyl acetate (800 ml). The organic layer was separated, washed with a 5% aqueous citric acid solution (400 ml), water (300 ml) and brine (300 ml), dried over anhydrous magnesium sulfate, and concentrated in vac- uo to afford the title compound (28.2 g, quant). |
|
With triethylamine; 1,1'-carbonyldiimidazole; In dichloromethane; at 20℃; for 1h; |
To a stirred solution of 1-[(tert-butoxy)carbonyl]azetidine-3-carboxylic acid (2.00 g, 9.94 mmol) and CDI (1.80 g, 10.9 mmol) in DCM (10 mL) were added Et3N (1.20 g, 11.93 mmol) and N,O-methoxy(methyl)amine hydrochloride (0.90 g, 14.91 mmol) at room temperature. The reaction solution was stirred at room temperature for 1 h. The resulting solution was diluted with water (30 mL) at room temperature and extracted with EA (3 x 30 mL). The combined organic layers were washed with brine (2 x 20 mL), dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EA (1/1) to afford tert-butyl 3- [methoxy(methyl)carbamoyl]azetidine-1-carboxylate as a light yellow oil (2.10 g, 78%): LCMS (ESI) calc’d for C11H20N2O4 [M + H - 56]+: 189, found 189; 1H NMR (300 MHz, CD3OD) δ 4.14-3.99 (m, 4H), 3.89-3.76 (m, 1H), 3.72 (s, 3H), 3.22 (s, 3H), 1.46 (s, 9H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping