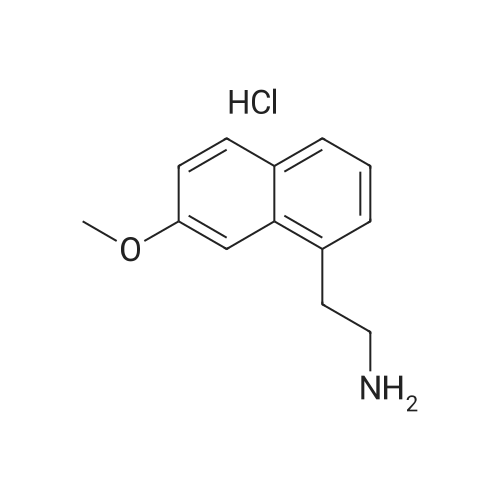
| 95% |
With ammonia; hydrogen; In ethanol; water; at 60℃; under 228015 Torr; for 12h;Autoclave; |
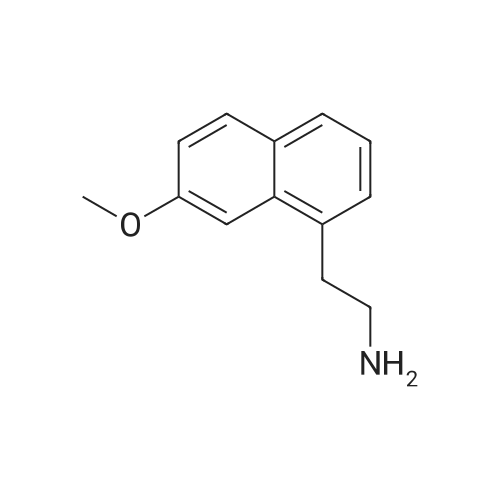
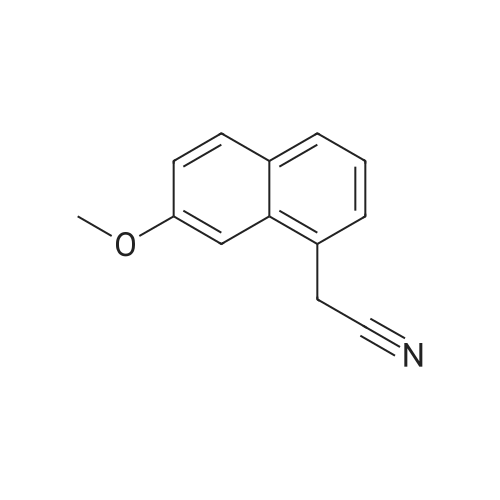
Reference example 6: The preparation of 2-(7-methoxy-1-naphthyl)ethylamine 56g 7-methoxy-1-naphthaleneacetonitrile, 120ml ammonia water, 332ml 95% ethanol, 20g Raney-Ni were added into autoclave. H2 was introduced after vacuuming it. The operation was repeated for 3 times. The reaction was stirred for 12 hours while H2 was introduced and the condition of 300 atm and 60C was maintained. After completion of the reaction, the reaction was kept overnight at room temperature. On the next day, it was vacuumed and N2 gas was introduced. The autoclave was opened up, and the reaction solution was filtered to remove the catalyst. The filtrate was vacuum evaporated until dry to obtain 56g light green oil. The content is measured as 96.95% by HPLC, and the yield is 95%. |
| 92% |
With hydrogen; ammonium hydroxide;nickel; In methanol; at 20℃; under 3102.97 Torr; for 20h; |
Step 7 2-(7-Methoxy-naphthalen-1-yl)-ethylamine: At ambient temperature and under a pressurized hydrogen atmosphere (60 psi), a mixture of (7-methoxy-naphthalen-1-yl)acetonitrile (1.6 g; 8.1 mmol), Raney nickel (0.5 g) in methanol (80 mL), and ammonium hydroxide (5 mL) was continuously stirred for about 20 hours. The catalyst was then removed by filtration and the filtrate was concentrated to give the title product as a yellow oil, which was used directly in the next step without further purification (1.5 g, yield 92%). 1H NMR (300 MHz, CD3OD): delta 3.00 (br. s, 2H), 3.20 (br. s, 2H), 3.93 (s, 3H), 7.12 (d, J=8.7 Hz, 1H), 7.24 (m, 3H), 7.63 (d, J=7.8 Hz, 1H), 7.73 (d, J=8.7 Hz, 1H). LC-MS: 202 (M+H)+. |
| 71.2% |
With ammonia; hydrogen; In ethanol; at 45℃; under 15001.5 Torr;Green chemistry; |
A hydrogenation reaction vessel was charged with <strong>[138113-08-3](7-methoxy-1-naphthyl)acetonitrile</strong> (10.0 g, 50.7 mmol), added to a 500 ml hydrogenreaction vessel, dissolved in 200 ml of anhydrous ethanol, and 0.95 g of Raney nickel wasadded to thehydrogenation reaction. kettle introducing gaseous ammonia, ammonia gas stream flatstable holding 0.1-5h; 1-5 times and then replaced by hydrogen, a sealed reactor; hydrogenation reactor was placed 45 C, 2.0MPa bar hydrogen pressureand stirred overnight under member; the reaction was monitored by TLC until complete; the reaction solution was decanted, suction filtered, rinsed with a little 95% ethanol; filtrate concentratedsolution has a pH, and dissolved in ethyl acetate, hydrochloric acid gas was introduced, the control of the mother liquor is 8.0 to 9.0, a large number of white precipitate heavyprecipitate Suction suspension was filtered off, the filter cake was washed with ethyl acetate, and the filter cake was dried at 50 C. to obtain an off-white solid, ie compound (II) 8.7 g, yield 71.2%, product properties: white powder.Purity: 99.52%. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping