* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With sulfuric acid; nitric acid; at 85℃;Cooling with ice;
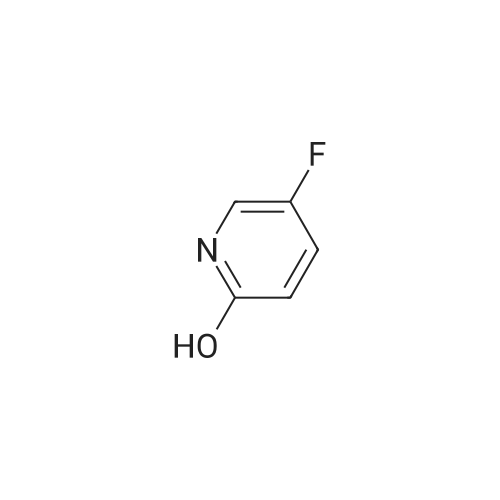
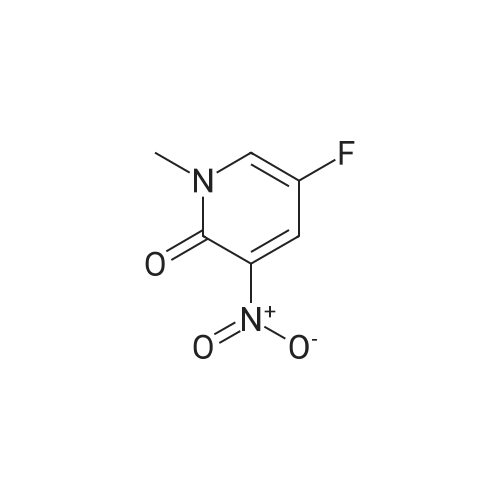
a) 5-Fluoro-3-nitropyridin-2-ol A mixture of concentrated sulphuric acid (1 mL) and fuming nitric acid (1 mL) was added dropwise to a stirred, cooled (ice-bath) mixture of 5-fluoropyridin-2-ol (1.20 g, 10.6 mmol) and concentrated sulphuric acid (2.7 mL). The mixture was warmed to ambient temperature and then to 85 C. After 2 hours, the mixture was cooled and poured onto ice-water. The precipitate was filtered and dried to give the title compound (0.72 g, 43%) as a yellow solid. LRMS (m/z): 157 (M-1)+. 1H NMR delta (300 MHz, DMSO-d6): 8.28 (s, 1H), 8.67 (s, 1H).
43%
With sulfuric acid; nitric acid; at 85℃; for 2h;Cooling with ice;
5-Fluoro-2-methoxypyridin-3 -amine a) 5-Fluoro-3-nitropyridin-2-olA mixture of concentrated sulphuric acid (1 mL) and fuming nitric acid (1 mL) was added dropwise to a stirred, cooled (ice-bath) mixture of 5-fluoropyridin-2-ol (1 .20 g, 10.6 mmol) and concentrated sulphuric acid (2.7 mL). The mixture was warmed to ambient temperature and then heated to 85 C. After 2 hours, the mixture was cooled and poured onto ice-water. The precipitate was filtered and dried to give the title compound (0.72 g, 43%) as a yellow solid.LRMS (m/z): 157 (M-1 )+.1H NMR delta (300 MHz, DMSO-d6): 8.28 (s, 1 H), 8.67 (s, 1 H).
40%
With sulfuric acid; nitric acid; at 28 - 65℃; for 2.75h;
To a solution of 5-fluoro-2-hydroxypyridine (200 mg, 1.77 mmol) in concentrated sulfuric acid (900 mul) was added, dropwise over 15 minutes, a premixed solution of concentrated sulfuric acid (900 mul) and fuming nitric acid (170 mul). The internal temperature rose by up to 280C. The reaction mixture was then heated at 650C for 2.5 hours. The cooled mixture was poured onto ice-water, and the pH of the mixture was adjusted to 2.5 with sodium carbonate. It was then extracted with ethyl acetate (2 x 25 ml). The aqueous layer was concentrated and extracted again with a mixture of tetrahydrofuran (25 ml) and ethyl acetate (25 ml). The organic layers were combined, dried over magnesium sulfate, filtered and concentrated under reduced pressure to yield the title compound (112 mg, 40%) as a solid. 1H NMR (400MHz, DMSO-D6): delta 8.22 (dd, 1 H), 8.60 (dd, 1 H); LRMS APCI" m/z 157 [M-H]'.
With sulfuric acid; nitric acid; In water; at 65 - 80℃; for 1h;
The solid from above containing (4-80) was divided in 4 batches and treated with H2SO4 and fuming HNO3 as shown below. The amounts used were: Compound 4-80 was dissolved in sulfuric acid (the larger amounts indicated above) at rt and then heated to 65 C. A preformed solution of fuming nitric acid and sulfuric acid (the smaller amount indicated above) was added dropwise. The temperature was kept between 65 C. and 80 C. (rxn is exothermic and although the bath is at 65 C., temperature goes higher, usually 75, sometimes 80 C.). After the addition was complete, the reaction mixture was heated at 65 C. for an additional hr. The reaction mixture was then cooled to rt and poured in a flask containing ice) (20 g of ice/gr compound, evolution of gas occurred). A solid precipitated out and it was collected by filtration (1HNM? showed 4-80 and something else (discarded)). [1493] The aqueous layer was extracted with AcOEt several times (3-5) and concentrated on a rotary evaporator under vacuum to afford a solid that was triturated with ether to afford 5-80 as a bright yellow solid. A total of 117 g of desired product was collected in the first crop (27% yield from diazonium salt). A portion did not crystallize: this oil was triturated with MeOH and Et2O to afford 3.6 g of 5-80; another precipitation from the mother liquid afforded an additional 6.23 g of the desired product 5-80 [1494] Total:117.0+3.6+6.23 =126.83. 30.4%). Yield for 3 steps (decomposition of diazonium salt; deprotection and nitration). [1495] Analytical data from Notebook: 53877-115: 1HNMR(delta, MeOD): 8.56-8.27 (dd, J=7.5, 3.3 Hz, 1H), 8.01 (d, J=3.3 Hz, 1H); LC/MS(M+1)+=158.9; rt=0.15 min. [1496] Note: A portion of the aqueous acidic solution was taken and neutralized with Na2CO3 until effervescence stopped and then it was extracted with AcOEtA different product was obtained. No desired product in these extracts.
With sulfuric acid; nitric acid; at 20 - 80℃; for 1h;
Intermediate 3 was dissolved in sulfuric acid (the larger amounts indicated above) at rt and then heated to 65 C. A preformed solution of fuming nitric acid and sulfuric acid (the smaller amount indicated above) was added dropwise. The temperature was kept between 65 C. and 80 C. (rxn is exothermic and although the bath is at 65 C., temperature goes higher, usually 75, sometimes 80 C.). After the addition was complete, the reaction mixture was heated at 65 C. for an additional hr. The reaction mixture was then cooled to rt and poured in a flask containing ice) (20 g of ice/gr compound, evolution of gas occurred). A solid precipitated out and it was collected by filtration (1HNM? showed intermediate 4 and something else (discarded)). The aqueous layer was extracted with AcOEt several times (3-5) and concentrated on a rotary evaporator under vacuum to afford a solid that was triturated with ether to afford intermediate 4 as a bright yellow solid. A total of 117 g of desired product was collected in the first crop (27% yield from diazonium salt). A portion did not crystallize: this oil was triturated with MeOH and Et2O to afford 3.6 g of intermediate 4; another precipitation from the mother liquid afforded an additional 6.23 g of the desired product intermediate 4. Total: 117.0+3.6+6.23=126.83. 30.4%). Yield for 3 steps (decomposition of diazonium salt; deprotection and nitration). Analytical data from Notebook: 53877-115: 1HNMR(delta, MeOD): 8.56-8.27 (dd, J=7.5, 3.3 Hz, 1H), 8.01 (d, J=3.3 Hz, 1H); LC/MS(M+1)+=158.9; rt=0.15 min.
With sulfuric acid; nitric acid; In water; at 65 - 80℃;
Compound 4-80 was dissolved in sulfuric acid (the larger amounts indicated above) at rt and then heated to 65 C. A preformed solution of fuming nitric acid and sulfuric acid (the smaller amount indicated above) was added dropwise. The temperature was kept between 65 C. and 80 C. (rxn is exothermic and although the bath is at 65 C., temperature goes higher, usually 75, sometimes 80 C.). After the addition was complete, the reaction mixture was heated at 65 C. for an additional hr. The reaction mixture was then cooled to rt and poured in a flask containing ice) (20 g of ice/gr compound, evolution of gas occurred). A solid precipitated out and it was collected by filtration (1HNM showed 4-80 and something else (discarded)). The aqueous layer was extracted with AcOEt several times (3-5) and concentrated on a rotary evaporator under vacuum to afford a solid that was triturated with ether to afford 5-80 as a bright yellow solid. A total of 117 g of desired product was collected in the first crop (27% yield from diazonium salt). A portion did not crystallize: this oil was triturated with MeOH and Et2O to afford 3.6 g of 5-80; another precipitation from the mother liquid afforded an additional 6.23 g of the desired product 5-80. Total: 117.0+3.6+6.23=126.83. 30.4%). Yield for 3 steps (decomposition of diazonium salt; deprotection and nitration). Analytical data from Notebook: 53877-115: 1HNMR(delta, MeOD): 8.56-8.27 (dd, J=7.5, 3.3 Hz, 1H), 8.01 (d, J=3.3 Hz, 1H); LC/MS(M+1)+=158.9; rt=0.15 min. Note: A portion of the aqueous acidic solution was taken and neutralized with Na2CO3 until effervescence stopped and then it was extracted with AcOEt A different product was obtained. No desired product in these extracts.
With benzyltrimethylammonium chloride; trichlorophosphate; In acetonitrile; at 80℃; for 6h;
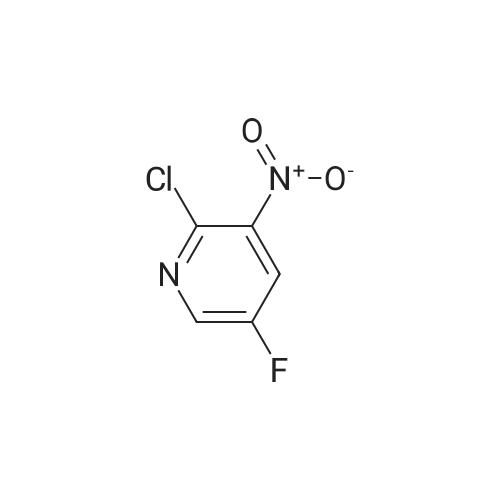
a) Synthesis of 2-chloro-5-fluoro-3-nitro-pyridine 5-fluoro-3-nitro-pyridin-2-ol (2 g, 12.7 mmol) and benzyltrimethyl ammonium chloride (1.17 g, 6.35 mmol) were dissolved in acetonitrile, and phosphorus oxychloride (3.5 ml, 38.1 mmol) was added thereto and stirred at 80 C. for 6 hours. The reaction mixture was cooled and poured into ice water to quench the reaction, and extracted with dichloromethane. The combined organic layer was washed with saturated saline solution, dried over anhydrous sodium sulfate (Na2SO4), filtered and evaporated under reduced pressure. The residue was purified by column chromatography on silica eluding with a solvent of dichloromethane:methanol=30:1. The fractions containing the product were collected and evaporated to obtain yellow liquid (1.57 g, 70%). 1H-NMR (CDCl3, 300 MHz); delta=8.56 (d, J=2.7 Hz, 1H), 8.40 (dd, J=6.5 Hz, 2.7 Hz, 1H). MS (ESI); 176.9 (M++1).
69%
With tetraethylammonium chloride; trichlorophosphate; In acetonitrile; at 90℃; for 24h;
PREPARATION 53 2-Chloro-5-fluoro-3-nitropyridine 5-Fluoro-3-nitro-pyridin-2-ol (1.00 g, 5.33 mmol) was dissolved in acetonitrile (40 mL) and tetraethylammonium chloride (2.10g, 12.65 mmol) was added. The mixture became clear, phosphorous oxytrichloride (1.94 g, 12.65 mmol) was added at room temperature and the mixture was heated at 90 oC for 24h. The reaction mixture was evaporated to dryness; the residue was taken up with water (100 mL) and extracted with ethyl acetate (2x100 mL). The organic layer was dried with sodium sulfate, filtered and evaporated to dryness. A yellow solid (0.90 g, 69%) was isolated, pure enough to perform the next synthetic step. LRMS (m/z): 177 (M+1)+
41%
With trichlorophosphate;N,N-dimethyl-formamide; at 110℃; for 18h;
A mixture of the pyridine of preparation 55 (105 mg, 0.66 mmol), phosphorus oxychloride (1 ml) and Lambda/,Lambda/-dimethyIformamide (10 mul, catalytic) was heated at 11O0C for 18 hours. The cooled reaction mixture was then concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane/acetonitrile as eluant (100:0 to 50:50 v/v) to yield the title compound (48 mg, 41%) as a solid. 1H NMR (400MHz, CDCI3): delta 8.03 (dd, 1 H), 8.55 (d, 1 H).
With trichlorophosphate; In 1,2-dimethoxyethane; at 110℃; for 5h;
In Step B, compound zz2' (500 mg, 3.16 mmol) was dissolved in phosphorous oxychloride (1.7 mL, 18.9 mmol) and dimethoxyethane at room temperature. The reaction was heated to 110 C. for 5 hours. The excess phosphorous oxychloride was then removed by concentrating the reaction mixture in vacuo. The residue was chromatographed on silica gel, eluted with chloroform (100%) to afford 176 mg of product zz3'.
With tetraethylammonium chloride; trichlorophosphate; In acetonitrile; at 20 - 90℃; for 24h;
5-Fluoro-3-nitro-pyridin-2-ol (1 .00 g, 5.33 mmol) was dissolved in acetonitrile (40 mL) and tetraethylammonium chloride (2.1 Og, 12.65 mmol) was added. The mixture became clear, phosphorous oxytrichloride (1 .94 g, 12.65 mmol) was added at room temperature and the mixture was heated at 90 C for 24h. The reaction mixture was evaporated to dryness; the residue was taken up with water (100 mL) and extracted with ethyl acetate (2x100 mL). The organic layer was dried with sodium sulphate, filtered and evaporated to dryness. A yellow solid (0.90 g, 69%) was isolated, pure enough to perform the next synthetic step.LRMS (m/z): 177 (M+1 )+
With N,N-dimethyl-formamide; phosphorus(V) oxybromide; at 20 - 110℃; for 3.08333h;
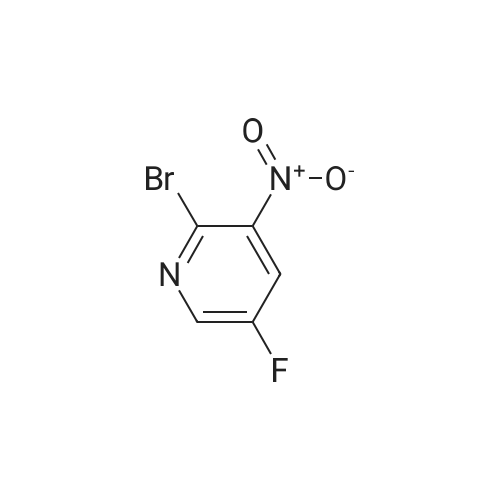
A total of 117 g of 5-80 was divided in 4 batches of 30 g×3 and 27 g×1 and treated with POBr3 (3 equiv.; 163 g×3 and 155 g×1) and a catalytic amount of DMF (15 ml) at rt (DMF was added carefully gas evolution). After 5 min. at room temperature, the solutions were heated at 110 C. for 3 hr. LC/MS showed starting material had been consumed. The reaction mixtures were allowed to cool to rt. The reaction flasks were placed in an ice bath; and then ice was added very slowly and carefully portionwise into the flask, gas evolution was due to HBr formation; the liquid and black solid that formed was poured into a beaker with ice. EtOAc was added and the mixture was then extracted several times with EtOAc. The organic layer was washed with saturated aq. NaHCO3; H2O and brine; dried over Na2SO4 and filtered. The product was dried in the pump overnight to provide 123 g of 6-80 as a brown solid (77% yield). [1498] Note: Reaction is completed within 1 h. [1499] 1HNMR(delta, CDCl3):8.52 (m, 1H), 7.93 (m, 1H).
77 - 97%
With phosphorus(V) oxybromide;N,N-dimethyl-formamide; at 20 - 110℃; for 3h;
A total of 117 g of intermediate 4 was divided in 4 batches of 30 g×3 and 27 g×1 and treated with POBr3 (3 equiv.; 163 g×3 and 155 g×1) and a catalytic amount of DMF (15 ml) at rt (DMF was added carefullygas evolution). After 5 min. at room temperature, the solutions were heated at 110 C. for 3 hr. LC/MS showed starting material had been consumed. The reaction mixtures were allowed to cool to rt. The reaction flasks were placed in an ice bath; and then ice was added very slowly and carefully portionwise into the flask, gas evolution was due to HBr formation; the liquid and black solid that formed was poured into a beaker with ice. EtOAc was added and the mixture was then extracted several times with EtOAc. The organic layer was washed with saturated aq. NaHCO3; H2O and brine; dried over Na2SO4 and filtered. The product was dried in the pump overnight to provide 123 g of intermediate 5 as a brown solid (77% yield). Note: Reaction is completed within 1 h. 1HNMR(delta, CDCl3):8.52 (m, 1H), 7.93 (m, 1H).
77%
With phosphorus(V) oxybromide;N,N-dimethyl-formamide; at 110℃; for 3h;
A total of 117 g of 5-80 was divided in 4 batches of 30 g×3 and 27 g×1 and treated with POBr3 (3 equiv.; 163 g×3 and 155 g×1) and a catalytic amount of DMF (15 ml) at rt (DMF was added carefully gas evolution). After 5 min. at room temperature, the solutions were heated at 110 C. for 3 hr. LC/MS showed starting material had been consumed. The reaction mixtures were allowed to cool to rt. The reaction flasks were placed in an ice bath; and then ice was added very slowly and carefully portionwise into the flask, gas evolution was due to HBr formation; the liquid and black solid that formed was poured into a beaker with ice. EtOAc was added and the mixture was then extracted several times with EtOAc. The organic layer was washed with saturated aq. NaHCO3; H2O and brine; dried over Na2SO4 and filtered. The product was dried in the pump overnight to provide 123 g of 6-80 as a brown solid (77% yield). Note: Reaction is completed within 1 h. 1HNMR(delta, CDCl3):8.52 (m, 1H), 7.93 (m, 1H).
With phosphorus(V) oxybromide; In DMF (N,N-dimethyl-formamide); at 110℃;
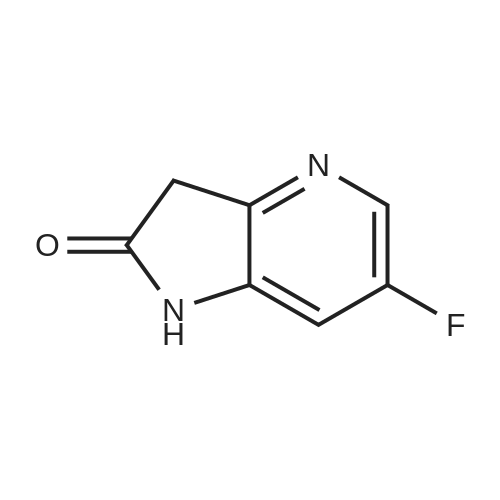
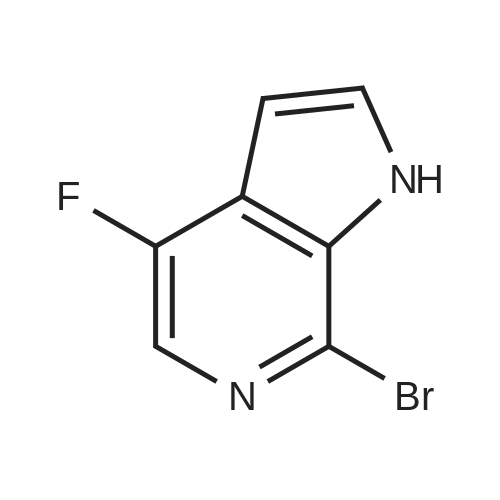
Intermediate 4,4-fluoro-7-bromo-6-azaindole, was prepared according to the following scheme: [CHEMMOL-00081] [0364] A) fuming HNO3, H2SO4; [0365] B) POBr3/DMF, 110 C.; [0366] C) vinylmagnesium bromide, THF, -78 C. -20 C. [0367] Intermediate 4 was isolated as a brownish solid. MS m/z: (M+H)+ calcd for C7H5BrFN2: 214.96; found 214.97. HPLC retention time: 1.28 minutes (column G).
With sulfuric acid; nitric acid; In water; at 85℃; for 1h;
In Step A, compound zzl' (1.2 g, 0.01 mol) was dissolved in sulfuric acid (2.7 mL) at room temperature. Premixed fuming nitric acid (1 mL) and sulfuric acid was added dropwise at 5-10 C. to the solution of compound zzl'. The reaction mixture was then heated at 85 C. for 1 hour, then was cooled to room temperature and poured into ice (20 g). The yellow solid precipitate was collected by filtration, washed with water and air dried to provide 1.01 g of compound zz2'.
With nitric acid;sulfuric acid;
Intermediate 4,4-fluoro-7-bromo-6-azaindole, was prepared according to the following scheme: [CHEMMOL-00081] [0364] A) fuming HNO3, H2SO4; [0365] B) POBr3/DMF, 110 C.; [0366] C) vinylmagnesium bromide, THF, -78 C. -20 C. [0367] Intermediate 4 was isolated as a brownish solid. MS m/z: (M+H)+ calcd for C7H5BrFN2: 214.96; found 214.97. HPLC retention time: 1.28 minutes (column G).
The combined extracts were dried (MgSO4) and concentrated to yield an additional amount of the title compound (1.71 g, 11%).
9
[ 136888-20-5 ]
[ 1257069-38-7 ]
Yield
Reaction Conditions
Operation in experiment
58.5%
With palladium 10% on activated carbon; hydrogen; In ethanol; at 20℃; for 2h;
5-Fluoro-3-nitropyridin-2-ol (10 g, 63.3 mmol) was dissolved in ethanol (300 mL), 1 0% Pd/C (1 .8 g) was added and the mixture wasstirred at room temperature under atmospheric pressure of hydrogen for2 h. Pd/C was removed by filtration and the solvent was evaporated invacuum to obtain the title compound as an off-white solid (7,5 g, 58.5mmol, Y=92%). LC-MS (M-H) = 129.0
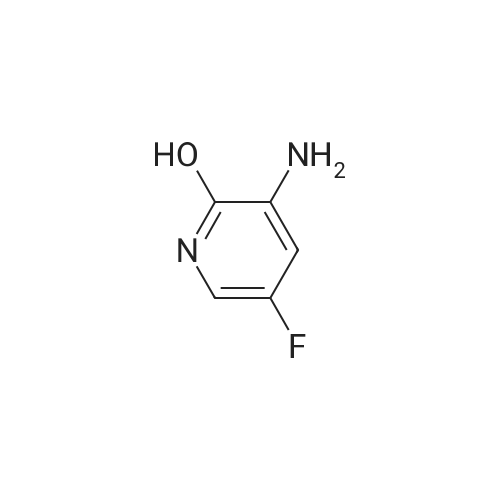
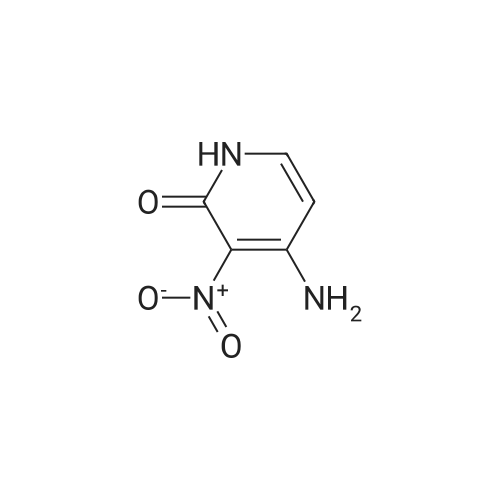
Step 4. 3-amino-5-fluoropyridin-2-olA mixture of <strong>[136888-20-5]5-fluoro-3-nitropyridin-2-ol</strong> (73 mg, 0.46 mmol; prepared according to procedure reported in WO 2006/114706) and iron (130 mg, 2.3 mmol), in ethanol (1.0 mL), acetic acid (0.76 mL), water (0.38 mL) and c. HCl (1 drop) was heated to 100 C. for 20 min. After cooling to RT, the solution was diluted with water (10 mL), filtered, and the filtrate was concentrated. The residue was then treated with saturated sodium bicarbonate solution to near pH 7, and the product was extracted with 6×40 mL of 20% isopropanol/DCM. The extracts were dried over sodium sulfate, filtered and concentrated and the product was used without further purification in the following step. LCMS (M+H)+: 129.0.
With dmap; triethylamine; In dichloromethane; at 25℃; for 16h;Inert atmosphere;
To a mixture of <strong>[136888-20-5]5-fluoro-3-nitropyridin-2-ol</strong> (24.2 g, 153 mmol, CAS 136888-20-3), tosyl-chloride (33.4 g, 176 mmol) in dichloromethane (1000 mL) was added at room temperature, under nitrogen atmosphere, triethyl amine (44 mL, 304 mmol). At the end of the addition, DMAP (3.7 g, 30 mmol) was added. The resulting mixture was stirred at 25C for 16 h. Dichloromethane (500 mL) was added and the mixture was successively washed with an aqueous solution IN of hydrochloric acid (2 X 500 mL) and brine (500 mL). The separated aqueous layer was extracted with CH2CI2 (400 mL). The combined organic layers were dried over Na2S04 and filtered through a silica pad (50 g). The filtrate was purified by flash chromatography (eluent: CH2CI2) to give intermediate 22-1 (34 g, 67 %).
With silver carbonate; In hexane; at 150℃; for 1h;Microwave irradiation;
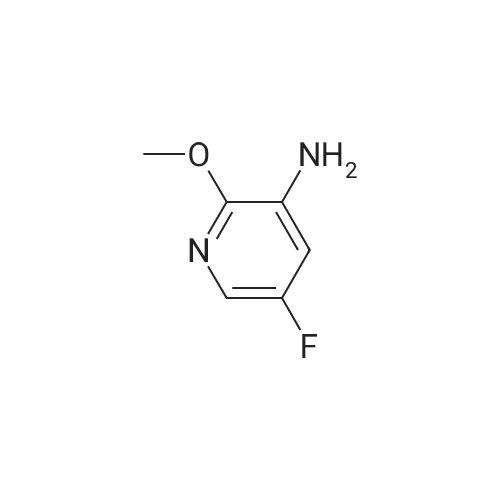
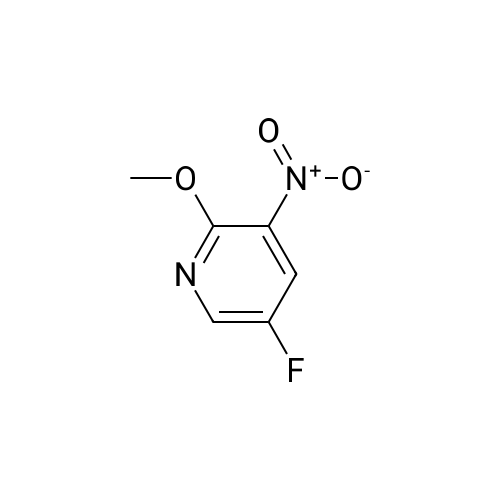
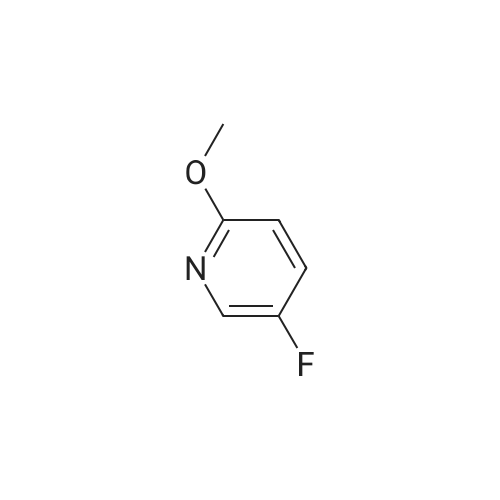
Method xxi-pyridyl amine 1: To a solution of <strong>[136888-20-5]5-fluoro-3-nitropyridin-2-ol</strong> (158 mg, 1 mmol) in n-hexane (1 mL) was added silver carbonate (331 mg, 1.2 mmol) and methyl iodide (0.2 mL, 2 mmol). The resulting mixture was stirred at 150 C under microwave irradiation (Power 250 W) for 1 hour. Solvent was removed and the residue was dissolved in ethyl acetate (2 ml) and washed with water (2x). Solvent was removed and the residue was purified on ISCO affording 5-fluoro-2-methoxy-3-nitropyridine (100 mg, 58%) as a colorless oil. 1H NMR (400 MHz, CDC13) delta 8.31 (d, J = 2.8 Hz, 1H), 8.09 (dd, J = 7.2, 2.8 Hz, 1H), 4.11 (s, 3H).
55%
With silver carbonate; In chloroform; at 20℃;
b) 5-Fluoro-2-methoxy-3-nitropyridine Iodomethane (1.97 mL, 31.7 mmol) was added to a suspension of <strong>[136888-20-5]5-fluoro-3-nitropyridin-2-ol</strong> (Preparation 5a, 0.500 g, 3.2 mmol) and silver(I) carbonate (1.04 g, 3.8 mmol) in chloroform (15 mL) and the mixture was stirred overnight at ambient temperature. The mixture was filtered through Celite, the filtrate was evaporated and the residue was purified by flash chromatography (2:1 hexanes/ethyl acetate) to give the title compound (0.300 g, 55%) as a white solid. LRMS (m/z): 173 (M+1)+. 1H NMR delta (300 MHz, CDCl3): 4.17 (s, 3H), 8.15 (dd, 1H), 8.37 (d, 1H).
55%
With silver carbonate; In chloroform; at 20℃;
b) 5-Fluoro-2-methoxy-3-nitropyridinelodomethane (1 .97 mL, 31.7 mmol) was added to a suspension of 5-fluoro-3- nitropyridin-2-ol (Preparation 5a, 0.500 g, 3.2 mmol) and silver(l) carbonate (1 .04 g, 3.8 mmol) in chloroform (15 mL) and the mixture was stirred overnight at ambient temperature. The mixture was filtered through Celite, the filtrate was evaporated and the residue was purified by flash chromatography (2:1 hexanes/ethyl acetate) to give the title compound (0.300 g, 55%) as a white solid. LRMS (m/z): 173 (M+1 )+.1H NMR delta (300 MHz, CDCI3): 4.17 (s, 3H), 8.15 (dd, 1 H), 8.37 (d, 1 H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping