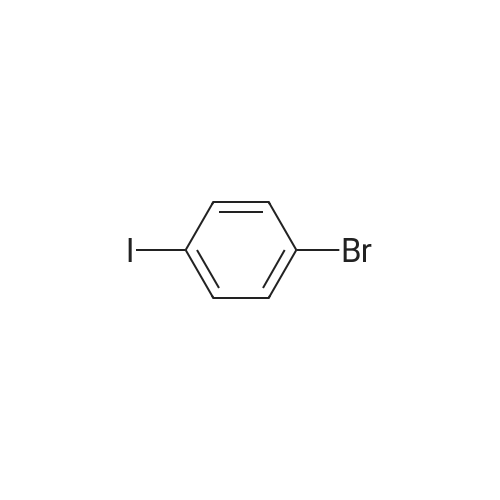
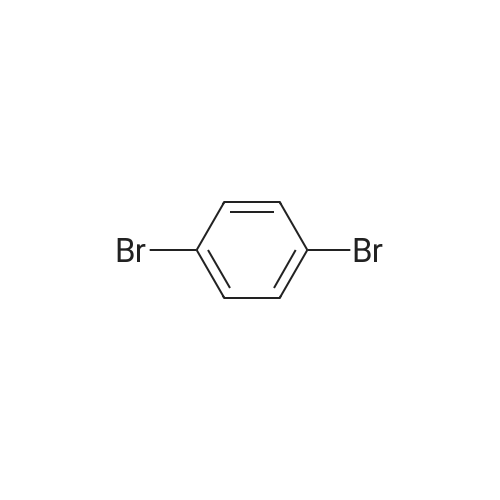
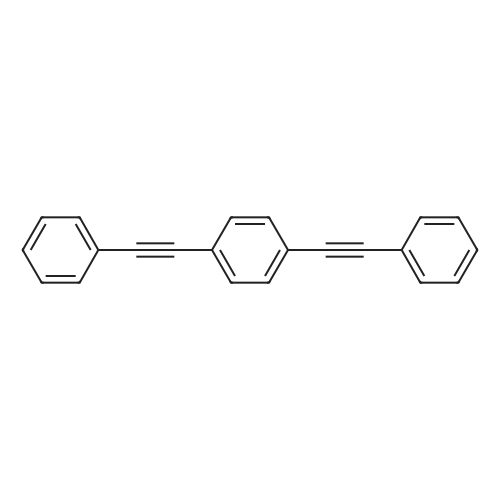
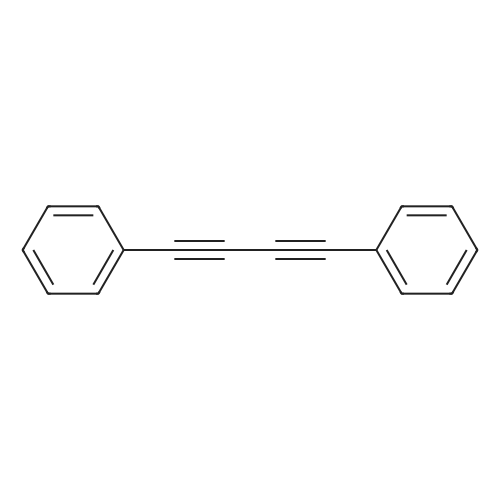
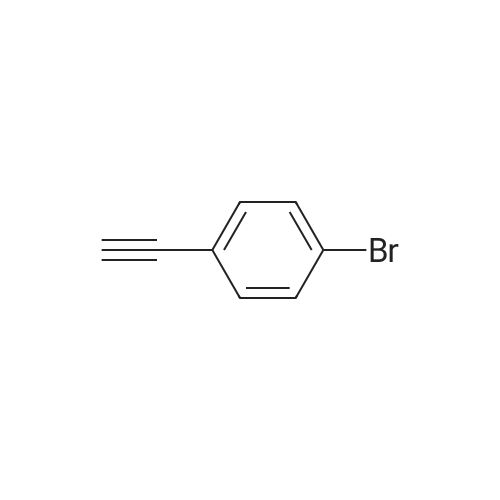
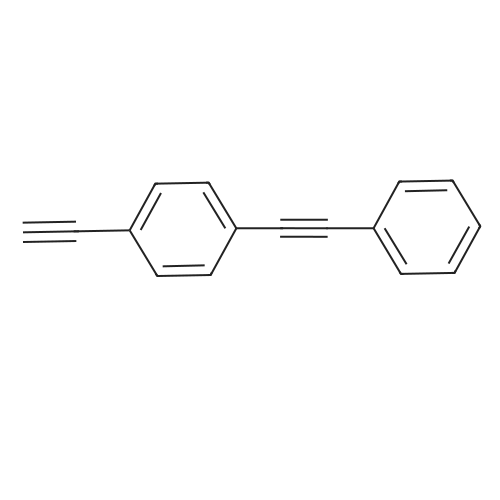
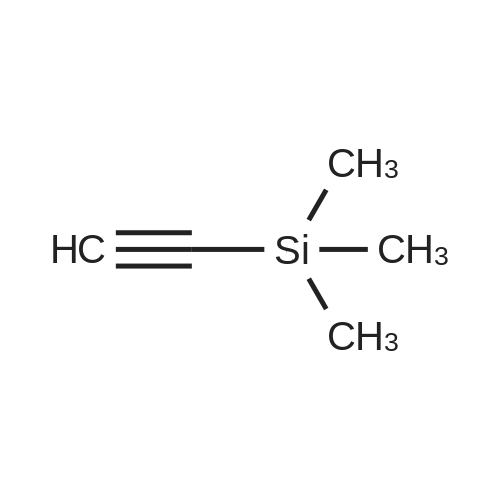
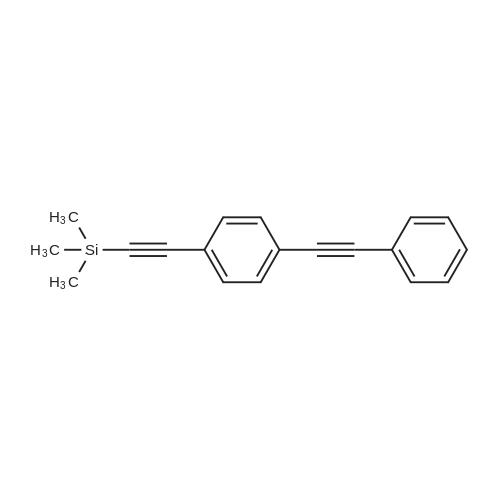
| 93% |
With pyridine; In neat (no solvent); at 20℃; for 3h; |
General procedure: Under air atmosphere, around-bottomed flask was charged with an aryl halide (1.0mmol), a terminal alkyne (1.0mmol), a base (1.0mmol), and the catalyst 2 (1mol%). The mixture was stirred at room temperature for 3h under aerobic conditions. After completion of the reaction, the mixture was filtered to recover the catalyst. The polymer was washed with water, methanol, and acetonitrile, vacuum-dried, and stored for a new run. After GC analysis, the solvent was removed under vacuum, and the crude product was subjected to silica gel column chromatography using CHCl3-CH3OH (97:3) as eluent to afford the pure product. |
| 92% |
With triethylamine; In water monomer; at 90℃; for 1.75h; |
General procedure: A mixture of aryl iodide (1.0mmol), terminal alkyne (1.5mmol), Et3N (2.0mmol), H2O (2mL), and the catalyst (0.012mmol, 1.2mol% Pd) was stirred at 90C under aerial conditions. The progress of the reaction was monitored by TLC. After completion, H2O was evaporated, and CHCl3 (10mL) was added to the reaction mixture, and the catalyst was recovered with centrifugation. The organic layer was washed with H2O (2×5mL), dried over anhydrous MgSO4, filtered and concentrated in vacuum. The crude product was further purified by preparative TLC (silica gel) using n-hexane as eluent to afford the desired product. All the products were characterized by IR, 1H, and 13C NMR spectroscopy. |
| 89% |
With potassium carbonate; In N,N-dimethyl-formamide; at 110℃; for 12h; |
General procedure: Into a conical flask (10 mL) a mixture of aryl halide (1.0 mmol), terminal alkyne (1.1 mmol), K 2 CO 3 (2 mmol, 0.28 g), Pd-NHC-MIL- 101(Cr) (1.0 mol %, 8 mg) and DMF (5.0 mL) were stirred at 110 C. The reactions were monitored by TLC. Stirring was continued until the consumption of the starting materials based on reaction time in Scheme 4 . After completion of the reaction, the mixture was fil- tered and cooled down to room temperature and then added wa- ter. The organic compound was extracted with ethyl acetate (3 ×5 mL) from the aqueous layer and dried over anhydrous Na 2 SO 4 , filtered, then concentrated in vacuum. The organic mixture was then purified by silica gel column chromatography employing n - hexane/ethyl acetate as the eluent, affording the pure correspond- ing product. |
| 88% |
With palladium diacetate; triethylamine; In water monomer; at 80℃; for 3h; |
General procedure: A solution of Pd(OAc)2 (0.34 mg, 0.0015 mmol) and ligandPEG-DAIL[BF4] (4.5 mg, 0.003 mmol) in deoxygenated H2O(2 mL) was stirred at room temperature for 30 min in air. Et3N(101 mg, 1 mmol), aryl halide (0.5 mmol), and terminal alkyne(0.75 mmol) were then successively added. The reaction mixturewas heated in an oil bath with magnetic stirring. Aftercooling to room temperature, the reaction mixture was added tobrine (15 mL) and extracted with diethyl ether (315 mL). Thesolvent was concentrated under vacuum and the product wasisolated by short column chromatography on silica gel. |
| 82% |
With copper oxide (I); Cs2CO3; In N,N-dimethyl-formamide; at 135℃; for 24h;Inert atmosphere; |
General procedure: A sealable vial equipped with a magnetic stir bar wascharged with Cs2CO3 (652 mg, 2.0 mmol) and Cu2O (7.0 mg,0.05 mmol) under a nitrogen atmosphere. The aperture of thevial was then covered with a rubber septum. Under a nitrogen atmosphere, aryl alkyne 1 (1.5 mmol), aryl iodide 2 (1.0 mmol), and DMF (0.5 mL) were added by syringe. The septum was then replaced by a screw cap containing a Teflon-coated septum, and the reaction vessel was placed at 135 C. After stirring at this temperature for 24 h, the heterogeneous mixture was cooled to r.t. and diluted with EtOAc (20 mL). The resulting solution was filtered through a pad of silica gel, then washed with EtOAc (20 mL), and concentrated to give the crude material which was then purified by column chromatography on silica gel to yield alkyne 3. |
| 82% |
With [1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); copper (I) iodide; triethylamine; triphenylphosphine; In tetrahydrofuran; at 20℃; for 12h; |
4-bromonium iodide (0.02 mol) 2.04 g (0.02 mol) of phenacetylene, 0.024 g (0.00034 mol) of diphenylphosphine palladium dichloride, 0.06 g (0.00027 mol ) Triphenylphosphine, 0.06 g (0.00037 mol) of CuI, 20 ml of tetrahydrofuran and 20 ml of triethylamine were reacted at room temperature for 12 hours.After completion of the reaction, the solid was removed by suction filtration, and the solvent was removed by steaming and separated by column chromatography to give a pale yellowish solid. |
| 80% |
With potassium hydroxide; In N,N-dimethyl-formamide; at 120℃; for 8h; |
General procedure: A mixture of phenylacetylene (1.2 mmol), aryl halide (1 mmol), KOH (2 mmol), DMF (5 mL) and Ni/Cu-MCM-41 (20 mg) are added into a 25 mL flask. The resulting mixture is stirred at 120 oC for appropriate reaction time. Progress of reactions is monitored by TLC. After completion of the reaction, the catalyst is separated by centrifugation. Then, the reaction mixture is extracted with ethyl acetate and water. The organic phase is dried by addition of MgSO4 and concentrated under reduced pressure. The residue is purified by column chromatography. |
| 79% |
With [Cu(N-(2-quinolynylmethylene)-1H-benzimidazole)(PPh3)2]PF6; potassium carbonate; In toluene; at 90℃; for 16h;Inert atmosphere;Catalytic behavior; |
General procedure: The Sonogashira coupling reaction of phenylacetylene with different aryl halides catalyzed by copper(I) complexes was carried out according to the procedure: 10mol% of copper(I) catalyst was added to 2mmol of respective aryl halide, 2.5mmol of phenylacetylene, 2mmol of K2CO3 in toluene and the reaction mixture was stirred for 16h at 90 under nitrogen. The reaction mixture was then cooled to room temperature and the solution was filtered to remove the precipitated base. The filtrate was concentrated and crude product was purified by column chromatography using ether:chloroform (9:1). The purified product obtained was characterized by elemental analyses, IR, 1H NMR and mass spectral studies. |
| 77% |
With tris-(dibenzylideneacetone)dipalladium(0); copper (I) iodide; triethylamine; triphenylphosphine; In tetrahydrofuran; at 20℃; for 4h;Inert atmosphere; |
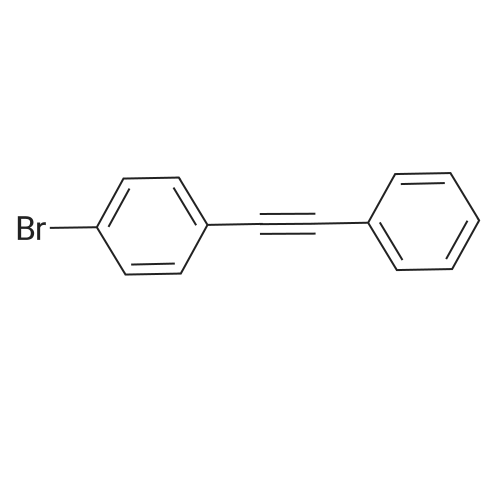
A mixture of 1-bromo-4-iodobenzene (2.540 g, 9.0 mmol), Pd2(dba)3 (137.6 mg, 0.15 mmol),triphenylphosphine (159.3 mg, 0.62 mmol), CuI (57.1 mg, 0.30 mmol), THF (48 mL) and Et3N (12mL) were placed in a round-bottom flask equipped with a magnetic stirring bar. After degassing thereaction mixture several times, ethynylbenzene (0.66 mL, 6.0 mmol) was added to the mixture. Thereaction was carried out at r.t. for 4 h with stirring. H2O was added to the reaction mixture. Theorganic layer was treated with 28% ammonia solution. The organic layer was extracted three timeswith CHCl3 and washed with brine. The organic layer was dried over MgSO4. MgSO4 was removedby filtration, and the solvent was removed by a rotary evaporator. The residue was purified bycolumn chromatography on SiO2 (hexane only) to affords S1 (1.3074 g, 4.6 mmol, 77%) as a whitesolid. Rf = 0.50 (hexane only). |
| 76% |
With C65H53CuN4P3(1+)*F6P(1-); potassium carbonate; In toluene; at 90℃; for 16h;Inert atmosphere; |
General procedure: The coupling of phenylacetylene with aryl halides catalyzedby these copper(I) complexes was carried outaccording to the following procedure: The copper(I) catalyst(10 mol %) was added to the respective aryl halide(2 mmol), phenylacetylene (2 mmol), and K2CO3(2 mmol) in toluene (10 ml), and the reaction mixture wasstirred for 16 h at 90 C under nitrogen. The reactionmixture was then cooled to room temperature, and thesolution was filtered to remove the precipitated base. Thefiltrate was concentrated to dryness, and the crude productwas purified by column chromatography using ether/chloroform(9:1). The purified product was then characterizedby elemental analyses, IR, 1H NMR and mass spectralstudies. |
| 75% |
With C53H46CuN4OP2(1+)*F6P(1-); potassium carbonate; In toluene; at 90℃; for 16h;Inert atmosphere; |
General procedure: The Sonogashira coupling reaction of phenylacetylene with aryl halides catalyzed by copper(I) complexes was carried out according to the procedure: 10 mol% of copper(I) catalyst was added to 2 mmol of respective aryl halide, 2.5 mmol of phenylacetylene, 2 mmol of K2CO3 in toluene and the reaction mixture was stirred for 16 h at 90 under nitrogen. The reaction mixture was then cooled to room temperature and the solution was filtered to remove the precipitated base. The filtrate was concentrated and crude product was purified by column chromatography using ether: chloroform (9:1). The purified product was then characterized by elemental analyses, IR, 1H NMR and mass spectral studies. |
| 74% |
With triethylamine; In water monomer; at 80℃; for 6h; |
General procedure: A 10 mL round-bottom flask was charged with iodobenzene (4a, 1 mmol, 1 eq.), phenylacetylene (7a, 1.5mmol, 1.5 eq.), triethylamine (3 mmol, 3 eq.), H2O (2 mL), and Pd catalyst (0.01 mmol). The flask was stirred at 80C in air. The reaction was monitored by TLC and GC. After the reaction was complete, the reaction mixture was cooled to room temperature and ethyl acetate (5mL) was added to the flask. Afterward, the catalyst was filtered and washed with water (10 mL) and ethyl acetate (10 mL). The aqueous phase was extracted three times with 30 mL EtOAc. The organic phases were collected together, dried over MgSO4, and filtered. The solvent was then evaporated under reduced pressure. The pure product was obtained via silica gel column chromatography with an eluent of EtOAc and hexane. The resulting product was analyzed by 1H NMR spectroscopy. |
| 70% |
With potassium carbonate; In ethanol; for 12h;Reflux; |
General procedure: in a typical procedure, the Sonogashira couplingreactions consist of aryl halide (0.5 mmol) in 3 mL of pure ethanol, phenylacetylene (0.75 mmol), K2CO3 (1 mmol) and 10 mg of the Pd-ZnFe2O4 catalyst(0.000764 mmol of Pd or 0.153 mol % of Pd). The reaction was carried outunder reflux condition for 11-14 h and was monitored by gas chromatography(GC). After completion, the reaction mixture was cooled to room temperatureand the catalyst (MNPs) was separated using an external magnet. The catalystwas then washed with ethanol, dried at 100 C for 3 h and preserved for nextcycle. The pure product was obtained using silica gel column chromatographyusing n-hexane-EtOAc as mobile phase. |
| 68% |
With C19H25CuN5(1+)*F6P(1-); potassium carbonate; In N,N-dimethyl-formamide; at 135 - 140℃;Sealed tube; |
General procedure: A 20mL scintillation vial was charged with a Teflon stir bar, copper complex (0.1mmol), 76 potassium carbonate (0.75mmol), aryl iodide (0.5mmol), 77 phenylacetylene (0.75mmol) in 5mL non-anhydrous DMF in air. The vial was sealed and placed in an oil bath with pre-adjusted temperature at 135-140C. After the allowed time, the reaction mixture was cooled down, diluted with 25-30mL ethyl acetate, and filtered through a pad of silica gel. The solvent was then removed under vacuum and the residue was purified by column chromatography using mixtures of hexane and ethyl acetate to obtain analytically pure product. |
| 65% |
With triethylamine; In ethanol; water monomer; at 25 - 30℃; for 3h;Green chemistry; |
General procedure: A mixture of aryl halid (0.125mmol), phenylacetylene(0.137mmol), NEt3(0.0625mmol) in 0.2ml H2O:EtOH(1:2) and catalyst (0.001g, 0.03mol%) was stirred at roomtemperature for the appropriate of time. The progress of thereaction was monitored by TLC. After completion the reaction,the catalyst was removed with an external magnet andwashed with EtOH, dried and used directly for a subsequentround of reaction without further purification. Then, desiredproduct (liquid phase) was extracted by plate chromatographyeluted with n-hexane/EtOAc (10:1). |
| 58% |
With triethylamine;bis(triphenylphosphine)palladium(II) chloride; copper monoiodide; In tetrahydrofuran; |
(i) Synthesis of (4-bromophenyl)phenylacetylene A synthesis scheme of (4-bromophenyl)phenylacetylene is shown in (B-1). Into a 1000-mL three-neck flask were added 28.3 g (0.10 mol) of p-bromoiodobenzene, 10.2 g (0.10 mol) of phenylacetylene, 701 mg (1 mmol) of bis(triphenylphosphine)palladium(II)dichloride, and 190 mg (1 mmol) of copper(I) iodide, and nitrogen substitution was carried out. Then, 350 mL of tetrahydrofuran and 18 mL of triethylamine were added thereto, and the mixture was stirred at room temperature for 12 hours. After the reaction, the reaction mixture was washed with a 3% hydrochloric acid aqueous solution, and an aqueous phase was extracted with ethyl acetate. The extract combined with an organic phase was washed with a saturated saline solution and then dried with magnesium sulfate. The mixture was filtered through Celite (produced by Wako Pure Chemical Industries, Ltd., Catalog No. 531-16855, the same product was used hereinafter), Florisil (produced by Wako Pure Chemical Industries, Ltd., Catalog No. 540-00135, the same product was used hereinafter), and alumina, and a solid obtained by the concentration of the filtrate was recrystallized with hexane to give 15 g of the target product as a solid at a yield of 58%. |
| 58% |
With [1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); copper (I) iodide; triethylamine; In tetrahydrofuran; at 20℃; for 12h;Inert atmosphere; |
p-bromo-iodobenzene 28.3g (0.10mol), phenylacetylene 10.2g (0.10mol), bis (triphenylphosphine) palladium (II) dichloride 701mg (1mmol), copper iodide (I) 190mg (1mmol) of into a 1000mL three necked flask,Nitrogen substitution, and then added to 350mL of tetrahydrofuran, 18mL of triethylamine was stirred at room temperature for 12 hours. After the reaction, the reaction mixture was washed with 3% hydrochloric acid solution and the aqueous layer is extracted with ethyl acetate. The combined extract and organic layer of a saturated aqueous sodium chlorideBy washing, it dried with magnesium sulfate. The mixture was Celite (Wako Pure Chemical Industries, Ltd., catalog number: 531-16855, hereinafter the same), Florisil (Wako Pure Chemical Industries, Ltd., catalog number: 540-00135, hereinafter the same), passed through an alumina filtered, and the solid obtained by concentration of the filtrate, the resulting recrystallized from hexane, the objectiveWater to give a solid 15g, yield 58%. |
| 55% |
With triethylamine;[1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); copper (I) iodide; In tetrahydrofuran; at 20℃; for 20h; |
In a 500 mL three-necked flask were placed 14 g (51 mmol) of p-bromoiodobenzene, 5.2 g (52 mmol) of phenylacetylene, and 98 mg (0.50 mmol) of copper(I) iodide. After the atmosphere in the flask was replaced with nitrogen, 200 mL of tetrahydrofuran and 9.0 mL of triethylamine were added to the flask, and the mixture was degassed by being stirred under reduced pressure. To this mixture was added 0.34 mg (0.50 mmol) of bis(triphenylphosphine)palladium(II) dichloride, and the mixture was stirred under a stream of nitrogen at room temperature for 20 hours. After a predetermined time, a 3% aqueous hydrochloric acid solution was added to the mixture, and an organic substance was extracted with ethyl acetate from the aqueous layer. The obtained extract was washed with a saturated aqueous sodium chloride solution together with the organic layer and then dried over magnesium sulfate. The mixture was subjected to suction filtration through Celite (produced by Wako Pure Chemical Industries, Ltd., Catalog No. 531-16855), Florisil (produced by Wako Pure Chemical Industries, Ltd., Catalog No. 540-00135), and alumina, and the filtrate was condensed to obtain a solid. The obtained solid was recrystallized with hexane; thus, 7.41 g of target light-brown powder was obtained with a yield of 55%. |
| 55% |
With triethylamine;[1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); copper (I) iodide; In tetrahydrofuran; at 20℃; for 20h; |
A synthetic scheme of (4-bromophenyl)phenylacetylene is shown in (A-1). In a 500 mL three-necked flask were placed 14 g (51 mmol) of p-bromoiodobenzene, 5.2 g (52 mmol) of phenylacetylene, and 98 mg (0.50 mmol) of copper(I) iodide. After the atmosphere in the flask was replaced with nitrogen, 200 mL of tetrahydrofuran and 9.0 mL of triethylamine were added to the flask, and the mixture was degassed by being stirred under reduced pressure. To this mixture was added 0.34 mg (0.50 mmol) of bis(triphenylphosphine)palladium(II) dichloride, and the mixture was stirred under nitrogen stream at room temperature for 20 hours. After a predetermined time, a 3 % aqueous hydrochloric acid solution was added to the mixture, and an organic substance was extracted with ethyl acetate from the aqueous layer. The obtained extract was washed with a saturated aqueous sodium chloride solution together with the organic layer and then dried over magnesium sulfate. The mixture was subjected to suction filtration through Celite (produced by Wako Pure Chemical Industries, Ltd., Catalog No. 531-16855), Florisil (produced by Wako Pure Chemical Industries, Ltd., Catalog No. 540-00135), and alumina, and the filtrate was condensed to obtain a solid. The obtained solid was recrystallized with hexane; thus, 7.4 g of target light-brown powder was obtained with a yield of 55 %. |
| 55.27% |
With [1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); copper (I) iodide; 1,8-diazabicyclo[5.4.0]undec-7-ene; In water monomer; toluene; at 20℃; for 18h;Inert atmosphere; |
General procedure: Under a nitrogen atmosphere,A 5 ml (17.67 mmol) of bromobenzene iodide was added to a 100 ml two-necked flask,Cuprous iodide (CuI) 336 mg (0.1 equ),1,8-diazabicycloundec-7-ene (DBU) 15.8 Ml (6 equ),Trimethyl ethynylsilane 1.25 Ml (0.5 equ),Phenanthroline Palladium dichloride 0.75 g (6%),Deionized water 0.125Ml (0.4equ), toluene 65mL;in room temperature,Stirring for 18 hours;After the reaction,Excessive DBU in hydrochloric acid neutralization system;Dry the toluene,Extracted twice with dichloromethane and water,Take the organic phase;The dichloromethane was removed by distillation under reduced pressure,Silica gel column purification,2 g of intermediate 1 was obtained in a yield of 34% |
| 52% |
With [1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); at 55℃; for 3h;Ionic liquid; Green chemistry; |
General procedure: In a 4 mL screw-cap vial, 0.5 mmol of corresponding iodoarenecompound, 1.5 equiv of phenylacetylene or propargyl alcohol,0.005 equiv PdCl2(PPh3)2, and 0.8 mL of ionic liquid weremixed and stirred at 55 C for 3 h. After cooling, the mixturewas partitioned between 5 mL of water and 5 mL of pentane.After separation, the aqueous phase was extracted subsequentlywith 2 × 5 mL of pentane. The combined organic phase waswashed with brine, dried over MgSO4, filtered, and the solventwas evaporated under reduced pressure (ca. 10 mmHg). Theoily residue was purified by chromatography on silica gel(Merck Silicagel 60 (0.063-0.200 mm) for column chromatography(70-230 mesh ASTM)) eluted with n-pentane/EtOAc.The purity of the isolated products was >98%. The detailed experimentalprocedure as well as the characterization of isolatedcompounds are provided in Supporting Information File 1. |
| 20% |
With 1,4-diaza-bicyclo[2.2.2]octane; manganese(III) triacetate dihydrate; at 70℃;Green chemistry; |
General procedure: In a 25 mL reaction tube, Mn(OAc)3.2H2O(10 mol%), DABCO (2.5 equiv.) and a stirring bar were added. Then iodobenzene(1 mmol), phenyl acetylene (1 mmol) and PEG-400 were injected by syringe. The reaction tube was closed and transferred to a 70 C oil bath for 19-24 hours. After the reaction completed, cool down the reaction mixture to room temperature. Water (2 mL) was added and the reaction mixture was extracted with ethyl acetate and then concentrated and purified by column chromatography. |
| 99%Chromat. |
With potassium carbonate; In water monomer; at 20℃; for 3h; |
General procedure: An aryl halide (1.0 mmol) and a terminal alkyne (1.2 mmol) were added to a mixture of PS-tazo-Pd(II) (0.001 mmol), K2CO3 (2.0 mmol), and water (3 ml) in a glass flask under vigorous stirring. The mixture was stirred at room temperature for 3 h under aerobic conditions. After completion of the reaction, the mixture was filtered to recover the catalyst. The polymer was washed with water and acetonitrile, vacuum dried, and stored for a new run. After GC analysis, the solvent was removed under vacuum, and the crude product was subjected to silica gel column chromatography using CHCl3-CH3OH (98:2) as eluent to afford the pure product. |
| 99%Chromat. |
With piperidine; In water monomer; at 20℃; for 3h; |
General procedure: An aryl halide (1.0 mmol) and a terminal alkyne (1.2 mmol) were added to a mixture of PS-dtz-Pd(II) (0.001 mmol), piperidine (2.0 mmol), and water (3 ml) in a glass flask under vigorous stirring. The mixture was stirred at room temperature for 3 h under aerobic conditions. After completion of the reaction, the mixture was filtered to recover the catalyst. The polymer was washed with water and acetonitrile, vacuum dried, and stored for a new run. After GC analysis, the solvent was removed under vacuum, and the crude product was subjected to silica gel column chromatography using CHCl3-CH3OH (97:3) as eluent to afford the pure product. |
| 95%Chromat. |
With triethylamine; In neat (no solvent); at 20℃; for 3h; |
General procedure: An aryl halide (1.0mmol) and a terminal alkyne (1.0mmol) was added to a mixture of PS-triazine-Pd(II) (0.001mmol) and base (1mmol) in a glass flask under vigorous stirring. The mixture was stirred at room temperature for 3h under aerobic conditions. Upon completion of the reaction, the reaction mixture was dissolved in chloroform (2mL). The palladium catalyst was separated from the mixture by filtration, washed with acetonitrile (10mL), and reused in the next run. Then to the chloroform solution was added toluene (1.0mmol) as the internal standard for GC analysis. After the analysis, the solvent was removed under vacuum, and the crude product was subjected to silica gel column chromatography using CHCl3-CH3OH (95:5) as eluent to afford the pure product. |
|
With copper (I) iodide; triethylamine; triphenylphosphine; palladium (II) chloride; In tetrahydrofuran; at 25 - 60℃; for 20h;Inert atmosphere; |
1-bromo-4-iodobenzene (mw=282.91 grams per mole (g/mol), v=0.5 moles (mol), m=141.46 grams (gr)) is dissolved in 1.5 liters (L) of a mixed solvent of triethylamine and tetrahydrofuran (volume to volume (v/v) of 1:1) in a 2 L round-bottomed flask to prepare a solution. Subsequently, the solution is purged with dry nitrogen gas for 1 hour (h). Subsequently, phenylacetylene (mw=102.14 g/mol, v=0.5 mol, m=51.1 gr), palladium(II) chloride (PdCl2, mw=177.33 g/mol, v=0.005 mol, m=0.89 gr), copper(I) iodide (CuI) (mw=190.45 g/mol, v=0.01 mol, m=1.9 gr), and triphenylphosphine (PPh3, mw=262.45 g/mol, v=0.02 mol, m=5.25 gr) are sequentially added to the solution. Then, the mixture is stirred at 25 C. for 8 h and at 60 C. for 12 h under a nitrogen atmosphere. Subsequently, 2-methyl-3-butyn-2-ol (mw=84.12 g/mol, v=0.6 mol, m=50.5 gr), palladium(II) chloride (PdCl2, mw=177.33 mol, v=0.0025 mol, m=0.45 gr), copper(I) iodide (CuI) (mw=190.45 g/mol, v=0.005 mol, m=0.95 gr), and triphenylphosphine (PPh3, mw=262.45 g/mol, v=0.01 mol, m=2.63 gr) are sequentially added thereto, and the mixture is stirred at 100 C. for 48 h under a nitrogen atmosphere. When the reaction is complete, the suspension is filtered to obtain a precipitate, and the precipitate is washed several times with a small amount of hot ethyl acetate. Then, the solvent is removed from the mother liquor under a reduced pressure, and the remaining solid is crystallized from ethyl acetate. The mother liquid is concentrated after the crystallization and dried at 60 C. under vacuum to obtain an intermediate I-1. The yield of the intermediate 1-1 is 88.3%. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping