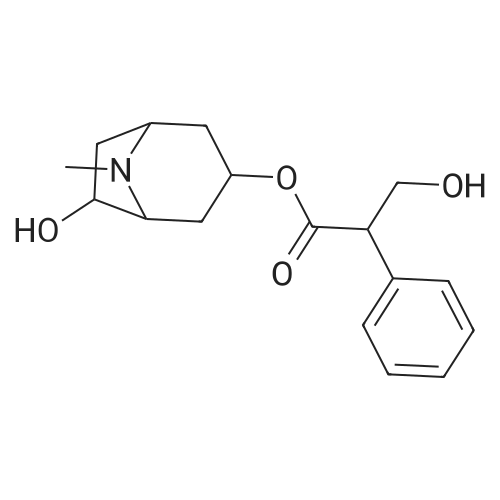
| 97.2% |
In N,N-dimethyl-formamide; acetone; at 20℃; |
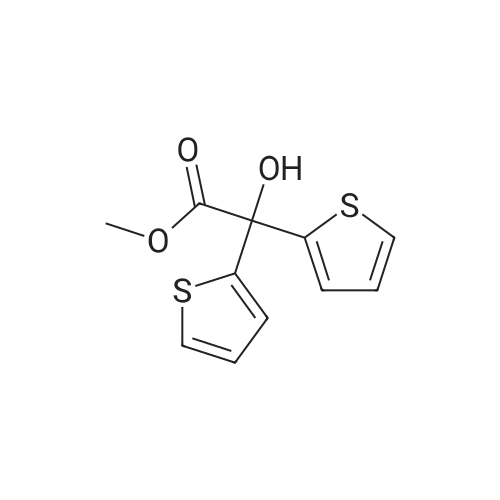
Example 11: Synthesis of tiotropium bromide A solution of 22% methyl bromide in acetone was prepared by scrubbing gas into a dark glass bottle containing 1.6 kg of acetone and placed on scales until obtaining 1.95 kg of methyl bromide solution in acetone.In a reaction flask were charged, in this order:scopine ester (3): 242.6g (0.6426 moles, 1 eq) DMF: 388 mL,acetone: 1370 mL(acetone/DMF ratio: about 3.5:1)The suspension, maintained under mechanical stirring, was heated to 40±5C until obtaining complete dissolution of the solid.The clear (pale yellow colored) solution was cooled to 20C and, keeping the flask immersed in a water bath, a 22.1% (w/w) solution of methyl bromide in acetone: 636 mL, equal to about 1.8 eq. of MeBr, was rapidly dropped, by addition funnel, on the clear solution.In a few hours of stirring at room temperature, a progressive precipitation of white solid was observed.The reaction mixture was maintained under stirring at room temperature. after about 4 days (90-100 h) a sampling was carried out for reaction control.The reaction mixture appeared as a heterogeneous suspension made up of clear mother waters and grainy and heavy solid, which tended to rapidly settle on the flask bottom.The solid was filtered, and flask and solid were washed over a filter with: 863 mL of acetone/DMF mixture (5:1), 431 mL of acetone/DMF mixture (5:1), 2x431 mL of acetone. The solid thus obtained was dried over the filter under Nitrogen current for 2-3h. At the end of the drying the solid appeared white and powdery.Crude dry Tiotropium bromide weight: 295.1 g (yield=97.2%) . |
| 97.3% |
In 2-methyltetrahydrofuran; N,N-dimethyl-formamide; at 0 - 30℃; |
<strong>[136310-64-0]N-demethyltiotropium</strong> (66 g; 0.17 mol) was dissolved in dimethylformamide (330 ml) and the solution was cooled to a temperature between 0 C and 5 C. A solution of bromomethane in 2-methyltetrahydrofuran (132 ml; 0.72 mol) was added and the reaction mixture stirred overnight at 0 C - 5 C. The content of <strong>[136310-64-0]N-demethyltiotropium</strong> in the reaction mixture was 3.6% by HPLC. Then the reaction mixture was heated up to a temperature between 10 C and 15 C and stirred at this temperature range over 2 hours. The content of <strong>[136310-64-0]N-demethyltiotropium</strong> in the reaction mixture decreased to 1.7%. The reaction mixture was heated to a temperature between 25 C and 30 C and stirred at that temperature range over 1 hour. The content of N- demethyltiotropium in the reaction mixture decreased to 1.0% by HPLC. 2- Methyltetrahydrofuran (594 ml) was added to the reaction mixture previously cooled to 0 C - 5 C, the suspension was stirred over 1 hour while maintaining the temperature between 0 C and 5 C, the product was filtered and washed with 2-methyltetrahydrofuran (297 ml) previously cooled to a temperature between.0 C and 5 C. The purity of the wet product was 99.48% and the content of <strong>[136310-64-0]N-demethyltiotropium</strong> was 0.33% (by HPLC). The wet product was re-s urried in dimethylformamide (297 ml) over 1 hour, was filtered, was washed with 2- methyltetrahydrofuran (297 ml) previously cooled' to 0 C - 5 C and dried. Crude tiotropium bromide (804 g; 97.3% of the theoretical yield) was obtained with a purity of .99.77% and with a residual content of <strong>[136310-64-0]N-demethyltiotropium</strong> of 0.16% (by HPLC) . - |
| 95% |
In acetone; acetonitrile; at 25 - 30℃; for 24h; |
Preparation of Anhydrous Tiotropium Bromide from Scopine di-(2-thienyl)glycolate Scopine di-(2-thienyl)glycolate (1 eq) was dissolved in acetone (35 vol) at 25-30 C. and methyl bromide in acetonitrile (50% w/w solution, 5 vol) was added. The mixture was stirred at 25-30 C. for 24 hours and the precipitated solid was filtered and washed with acetone (5 vol). The solid was dried at 60 C. under vacuum to afford the product as a white solid. The crude anhydrous tiotropium bromide obtained was found to have the XRPD, DSC and TGA spectra shown in , 2 and 3 respectively. Molar yield=95% HPLC purity=99.5-99.7% Purification of Anhydrous Tiotropium Bromide |
| 88% |
In DMF (N,N-dimethyl-formamide); at 20 - 30℃; for 60h; |
Methyl bromide (5.1 kg) is piped into the scopine ester solution obtained by the method described above at 20 C. The contents of the apparatus are stirred at 30 C. for about 2.5 days. 70 L of DMF are distilled off at 50 C. in vacuo. The solution is transferred into a smaller apparatus. It is rinsed with DMF (10 L). Additional DMF is distilled off at 50 C. in vacuo until a total amount of distillate of about 100 L is obtained. This is cooled to 15 C. and stirred for 2 hours at this temperature. The product is isolated using a suction filter drier, washed with 15 C. cold DMF (10 L) and 15 C. cold acetone (25 L). It is dried at max. 50 C. for max. 36 hours in a nitrogen current. Yield: 13.2 kg (88%); [0060] Melting point: 200-230 C. (depending on the purity of the starting product); [0061] The crude product thus obtained (10.3 kg) is added to methanol (66 L). The mixture is refluxed to dissolve it. The solution is cooled to 7 C. and stirred for 1.5 h at this temperature. The product is isolated using a suction filter drier, washed with 7 C. cold methanol (11 L) and dried for max. 36 h at about 50 C. in a nitrogen current. [0062] Yield: 9.9 kg (96%); [0063] Melting point: 228 C. (determined by TLC at a heating rate of 10 K/min). [0064] If desired, the product thus obtained may be converted in the crystalline monohydrate of tiotropium bromide. The following method may be used. [0065] 15.0 kg of tiotropium bromide are added to 25.7 kg of water in a suitable reaction vessel. The mixture is heated to 80-90 C. and stirred at constant temperature until a clear solution is formed. Activated charcoal (0.8 kg), moistened with water, is suspended in 4.4 kg of water, this mixture is added to the solution containing tiotropium bromide and rinsed with 4.3 kg of water. The mixture thus obtained is stirred for at least 15 min. at 80-90 C. and then filtered through a heated filter into an apparatus which has been preheated to an outer temperature of 70 C. The filter is rinsed with 8.6 kg of water. The contents of the apparatus are cooled to a temperature of 20-25 C. at a rate of 3-5 C. every 20 minutes. Using cold water the apparatus is cooled further to 10-15 C. and crystallisation is completed by stirring for at least another hour. The crystals are isolated using a suction filter drier, the crystal slurry isolated is washed with 9 L of cold water (10- 15 C.) and cold acetone (10-15 C.). The crystals obtained are dried at 25 C. for 2 hours in a nitrogen current. [0066] Yield: 13.4 kg of tiotropium bromide monohydrate (86% of theory). [0067] Melting point: 230 C. (determined by TLC at a heating rate of 10 K/min). |
|
In acetonitrile; at 22℃; for 12 - 64h;Product distribution / selectivity; |
Example 2: Preparation of crude Tiotropium bromide <n="29"/>[0109] 0.52 g of N-demethyl tiotropium (1.39 mmol) was suspended in 5.23 iriL of CH3CN under nitrogen.[0110] 1.35 g of CH3Br 50% w/w solution in CH3CN (0.0071 mol) were loaded, and the suspension was left under stirring at 220C for 12 hours. The product was filtered and washed with ImI of CH3CN.[0111] 572 mg of wet Tiotropium bromide were obtained (HPLC purity 99.89%, dithienylglycolic acid not detected).; Example 3 : Preparation of Tiotropium bromide[0112] 4.96g of N-demethyl tiotropium (13.2 mmol) were loaded in a flask under nitrogen with 49.6mL of CH3-CN. A suspension was obtained. 12.61g of CH3Br 50% w/w -CH3CN solution- (0.066 mol) were loaded.[0113] The suspension was left under stirring at 22C for 64 hours. The product was filtered and washed with 2mL of CH3CN. [0114] 6.93g of wet Tiotropium were obtained, and dried under vacuum at 45C for 22h (residual pressure 4 mbar) . 5.9 g of dry product (purity 99.8%, dithienylglycolic acid-not detected) were obtained. |
|
In acetone; acetonitrile; at 25 - 30℃; for 24h; |
Preparation of anhydrous tiotropium bromide from scopine di-(2-thienyl)glycolateScopine di-(2-thienyl)glycolate (1 eq) was dissolved in acetone (35 vol) at 25-30C and methyl bromide in acetonitrile (50% w/w solution, 5 vol) was added. The mixture was stirred at 25-3O0C for 24 hours and the precipitated solid was filtered and washed with acetone (5 vol). The solid was dried at 60C under vacuum to afford the product as a white solid. The crude anhydrous tiotropium bromide obtained was found to have the XRPD, DSC and TGA spectra shown in Figures 1, 2 and 3 respectively.Molar yield = 95%HPLC purity = 99.5-99.7%Purification of anhydrous tiotropium bromideCrude anhydrous tioteopium bromide (1 eq) was taken in DMSO (2 vol) and stirred for 1 hour at 25-3O0C. Acetone (25 vol) was slowly added and the mixture was chilled to 0-5C and stirred at 0-5C for 30 minutes. The solid was filtered and washed with acetone (3 vol) and dried under vacuum at 600C for 12 hours. The purified anhydrous tiotropium bromide obtained was found to have the XRPD, DSC and TGA spectra shown in Figures 1, 2 and 3 respectively.Molar yield = 98%HPLC purity > 99.9%The crude and purified samples of anhydrous tiotropium bromide prepared in the above examples were found to be substantially pure polymorphically with no levels of other- forms detected (>99.7% polymorphically pure, as measured by XRPD). The purified anhydrous tiotropium bromide prepared was also found to be very stable chemically and polymorphically with no conversion over time to other polymorphs. The stability of the sample was tested by subjecting the sample to accelerated stability conditions (400C + 2C temperature and 75% +/- 5% relative humidity) for 6 months. The chemical purity (measured by related substances and purity assays by HPLC) and polymorphic purity (measured by XRPD, DSC and TGA) were monitored for 6 months and the sample was found to be chemically and polymorphically stable even after 6 months under accelerated stability conditions.The XRPDs were recorded on a Bruker D8 Advance Diffractometer, using Cu Kalphal radiation as the X-ray source and LynxEye as the detector, with a 2theta range of from 3 to 50, a step-size of 0.05 and a time/step of lsec.The DSCs were recorded on a Perkin Elmer Pyris 6 Instrument over a temperature range of from 250C to 25O0C at a rate of heating of 10C/min. The TGAs were recorded on a Perkin Elmer Pyris 1 Instrument over a temperature range of from 250C to 250C at a rate of heating of 10C/min. |
| 2.65 g |
In dichloromethane; acetonitrile; at 20℃; for 48h; |
2.0 g of scopine di(2-thienyl)glycolate (5.3 mmol) was dissolved in 20 ml of a mixture of dichloromethane (8 ml) and acetonitrile (12 ml) at 55C. The solution was cooled down to 33C and 5.46 g of 55% MeBr in acetonitrile (5.1 equivalents) was added. The solution was stirred and left to cool freely without stirring still for 48 hours. The formed crystalline product was filtered, washed with 30 ml of dichloromethane, and dried in a vacuum oven at 35C for 6 hours. 2.65 g of white crystals were obtained,HPLC purity 99.60%. The content of solvents (determined by GC): dichloromethane 69,000 ppm, acetonitrile 8,200 ppm. |
| 30.8 g |
In dichloromethane; acetonitrile; at 20 - 33℃; for 19h; |
23.6 g of the scopine ester I (39.7 mmol) was dissolved in 230 ml of a mixture of dry dichloromethane (90 ml) and dry acetonitrile (140 ml) at 55C, then the solution was cooled to 33C and 54.0 g of 55% MeBr in dry acetonitrile (5.0 equivalents) were added. The solution was left to cool freely and then it was stirred at the room temperature for 19 hours. The resulting crystalline product was filtered, washed with 900 ml of dichloromethane and dried in a vacuum drier (5 kPa) at 35C for 17 hours. 30.8 g of white crystals were obtained, HPLC purity 99.74%. The content of solvents (measured with gas chromatography): dichloromethane 67000 ppm, acetonitrile 4300 ppm. |
| 30.8 g |
In dichloromethane; acetonitrile; at 20 - 33℃; for 19h; |
Example 9 (Reference example reproducing the method of patent EP0418716) 23.6 g of the scopine ester I (39.7 mmol) was dissolved in 230 ml of a mixture of dry dichloromethane (90 ml) and dry acetonitrile (140 ml) at 55C, then the solution was cooled to 33C and 54.0 g of 55% MeBr in dry acetonitrile (5.0 equivalents) were added. The solution was left to cool down freely, and subsequently stirred at the room temperature for 19 hours. The resulting crystalline product was filtered, washed with 900 ml of dichloromethane and dried in a vacuum drier (5 kPa) at 35 C for 17 hours. 30.8 g of white crystals were obtained, UPLC purity 99.74%. Solvent content (determined by gas chromatography): dichloromethane 67000 ppm, acetonitrile 4300 ppm. |
| 15.51 g |
In 1-methyl-pyrrolidin-2-one; at 20℃; |
Example 3 Preparation of Tiotropium Bromide (1) in NMP To a solution of 13.2 g (39.1 mmol) desmethyl-tiotropium (4) in 30 mL NMP, 16.5 mL (115 mmol, 2.93 eq.) of a 1:1 (w/w) solution of methyl bromide in NMP were added. The mixture was stirred overnight at room temperature, whereupon a dense suspension formed. After addition of 20 mL acetonitrile, the suspension was filtered, washed with 20 mL acetonitrile, and dried with high vacuum overnight at 30 C., yielding 15.51 g of off-white powder. Residual solvents were detected by GC analysis. The XRPD pattern complied with the one shown in . 1H-NMR (300 MHz, d6-DMSO): 7.52 (dd, J=5.0 Hz, 1.1, 2H), 7.41 (s, 1H), 7.13 (dd, J=3.6, 1.1 Hz, 2H), 7.01 (dd, J=5.0, 3.7 Hz, 2H), 5.12 (t, J=5.8 Hz, 1H), 4.13 (bd, J=5.8 Hz, 2H), 3.50 (s, 2H), 3.25 (s, 3H), 3.05 (s, 3H), 2.8-2.6 (m, 2H), 1.93 (s, 1H), 1.87 (s, 1H). 13C-NMR (75.5 MHz, d6-DMSO): 170.2, 147.1, 127.3, 126.7, 126.3, 76.8, 65.0, 64.2, 56.5, 54.1, 47.6, 28.7. |
|
In chloroform; acetonitrile; at 20℃; for 48h; |
A solution of compound 5 (1 equiv) in acetonitrile and chloroform mixture (10 mL, 1:1) was treated with bromomethane gas (4 equiv). The resulting mixture was stirred at room temperature. After 48h, the solution was filtered and the residue was dissolved in water (2.5 volumes) and heated up to 90 - C until it becomes clear solution, which was then treated with activated charcoal. The solution was filtered, washed with 0.5 mL of water and cooled to 5 · C, then filtered and washed with diethyl ether/acetone to afford the pure monohydrate (6) as a white solid. The product (6) is isolated as a monohydrate as evidenced by the presence of moisture content between 2.5%-4.0% measured by Karl Fisher test as per EP [tiotropium bromide monohydrate]. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping