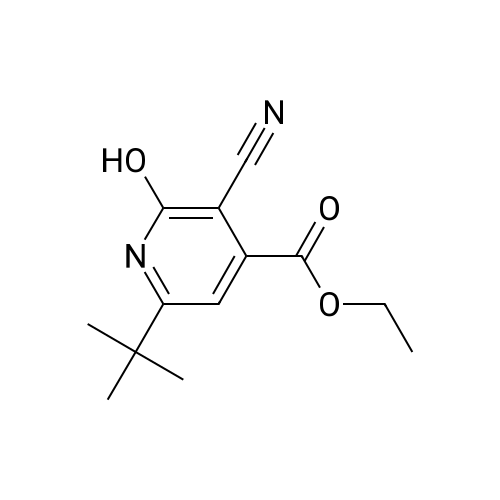
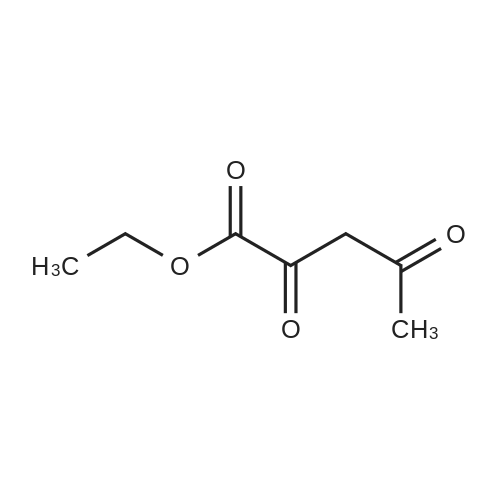
| 93.3% |
|
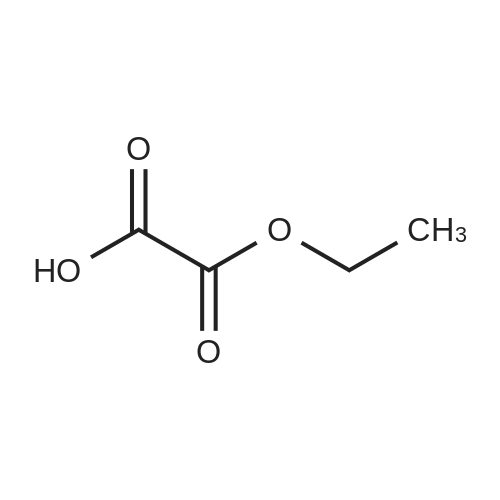
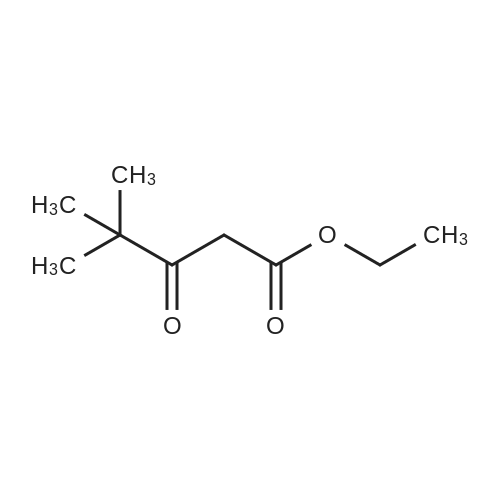
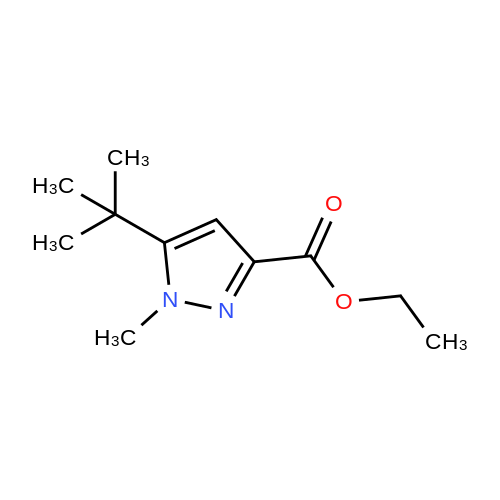
To a solution of sodium ethoxide (740mmol of sodium in 45OmL ethanol), a mixture of methyl tert-bxAy ketone (740mmol) and diethyl oxalate (820mmol) are added dropwise at room temperature and stirred for Ih at 78C. On complete reaction, the mixture is allowed to cool to room temperature and it is poured onto cold aqueous hydrochloric acid (5M, 800 mL). Following extraction with methyl tert-butyl ether (300 mL x 3), the combined organic phases are washed with saturated aqueous sodium chloride (500 mL) and dried over anhydrous sodium sulfate. After filtration, removal of the solvent under reduced pressure affords the product as a red liquid. Compound wt: 14Og, Yield: 93.3%.[00292] MS (ES+): 199 (M+l). NMR (1H, CDCl3): 6.5 (IH, s, CH); 4.3 (2H, q, OCH2); 1.35 (3H, t, CH3); 1.15 (9H, s, 3xCH3). |
| 74.6% |
With sodium; In ethanol; at 0 - 20℃; |
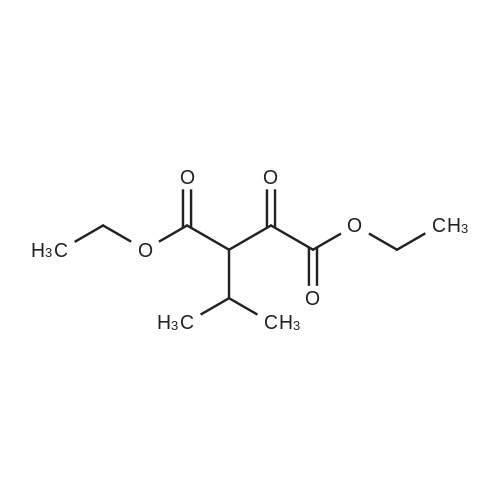
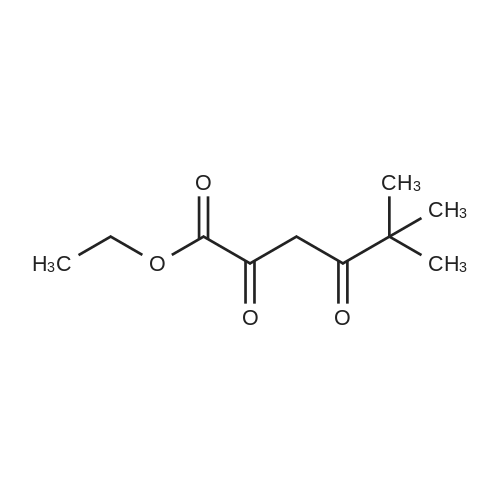
General procedure: A mixture of ketone 15(0.087 mol), diethyl oxalate 16(0.087mol),thinly sliced sodium (0.047mol) in ethanol (55 mL) was first stirred at 0 and then stirred over night at room temperature. After complication, the mixture was acidified(pH3.0,with 20% H2SO4), filtered and extracted with dichloromethane.The organic phase was dried and concentrated under vacuum to obtainan yellow or orange liquid 17a-e. The 1,3-diketone 17a-e(0.025mol) was dissolved in methanol (10mL),then added dropwise to a cooled solution(0C) of hydrazinobenzene(0.025 mol) in methanol (30mL). The reaction was stirred for an hour at room temperature, refluxed for 2h,and the solvent was evaporated under reduced pressure. The residual liguid was purified by column chromatography (a5% gradient of ethyl acetate in hexanes over a column of silica gel) to afford immediate 18a-e.To obtain the acid 19a-e, a solution of 18a-e(0.007 mol) was saponified through adding7ml 6 mol/L NaOH and stirring at 80Cfor 2 h.The mixture was acidified (pH1-2) with concentrated hydrochloric acid and filtered to afford 19a-e.To a solution of 19a-e(0.010mol) in 15ml DMF, NCS (0.010mol) was added.The reaction mixture was heated at 90C for 1hand then added to ice water(50ml), and filtered t oobtain 4-chlorosubstituted carboxylic acids 20a-e. The amide derivatives 21-38 were prepared through the acyl chlorides derived from 19 a-e or 20a-e. A solution of 19a-e or 20a-e(0.004mol) in thionyl chloride(10mL) was refluxed for 5 h and then concentrated under vacuum. The crude acylchloride was added dropwise to a cooled solution(0C) of substituted aniline (0.004mol) and TEA (0.008mol)in dichloromethane (10mL). The mixture was stirred overnight at room temperature, and then purified on a column of silica using a gradient of ethylacetate in hexanes to afford the pure products.The yields of imtermediate 17a-e, 18a-e, 19a-e and 20a-e are listed intable 1S. |
| 74.6% |
With sodium; In ethanol; at 0 - 20℃; |
Pinacolone 1 (0.087 mol) and diethyl oxalate 2 (0.087 mol) were added dropwise to a solution of ethanol (55 mL) containing thinly sliced sodium (0.047 mol) at 0 C. The mixture was stirred overnight at room temperature. Next morning, the mixture was acidified (pH 3.0, with 20% H2SO4) and filtered to remove the formed solid. The filtrate was extracted with dichloromethane, dried, and concentrated under vacuum to yield an orange red viscous liquid 3 (12.98 g; 74.6% y). A solution of the 1,3-diketone 3 (0.025 mol) in methanol (10 mL) was added dropwise to a cooled solution (0 C) of hydrazinobenzene (0.025 mol) in methanol (30 mL). The mixture was warmed to room temperature by stirring for an hour and then refluxed for 2 h. The resulting cooled mixture was concentrated under vacumm and the pyrazole ester 4 was obtained after purification by column chromatography (a 5% gradient of ethyl acetate in hexanes over a column of silica gel). To saponify the ester, a solution of 4 (0.007 mol) was combined with an aliquot of 6 mol/L NaOH (aq) (7 mL) and the mixture was stirred at 80 C. Ice water (50 mL) was added at the end of 2 h and the mixture was acidified (pH 1-2) with concentrated HCl. The formed solid was collected by filtration and the filter-cake was dried. The carboxylic acid was purified by recrystallization (methanol:water, 1:1) to afford 5 (3.57 g; 83.2% yield). The amide derivatives 6a-p were prepared through the acylchlorides derived from 5. A solution of 5 (0.004 mol) in thionyl chloride (10 mL) was refluxed for 5 h [11] and then concentrated under vacuum. The formed crude acyl chloride was added dropwise to a cooled solution (0 C) of substituted aniline (0.004 mol) and TEA (0.008 mol) in dichloromethane (10 mL). The resulting mixture was stirred overnight at room temperature to produce the crude product, which was purified on a column of silica using a gradient of ethyl acetate in hexanes to afford the pure products 6a-p. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping