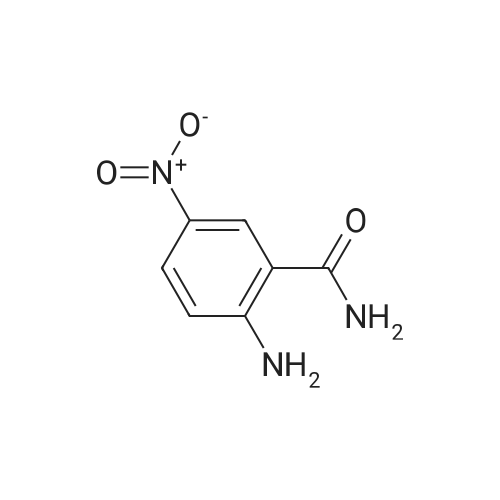
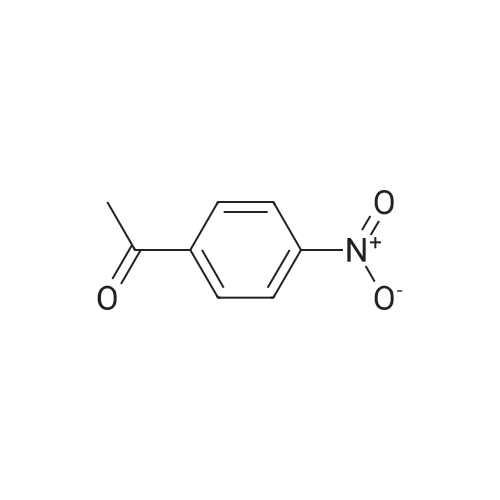
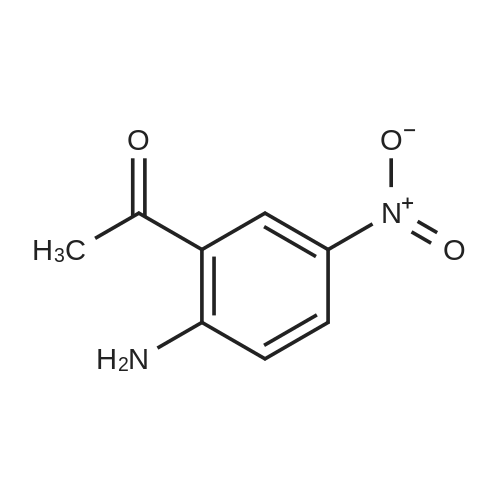
| 95% |
With sodium tetrahydroborate; In methanol; at 0℃; for 1h; |
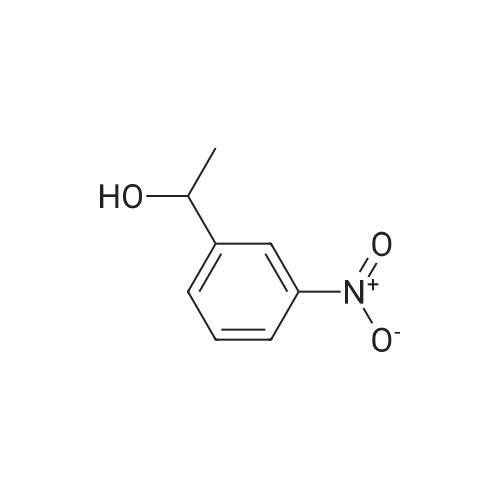
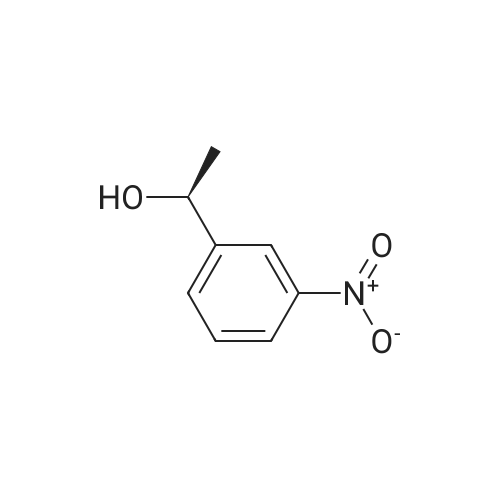
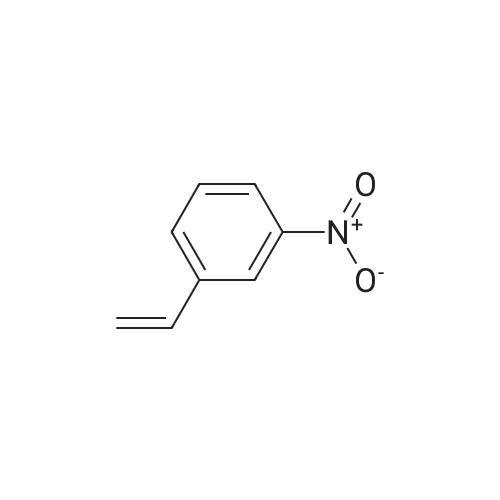
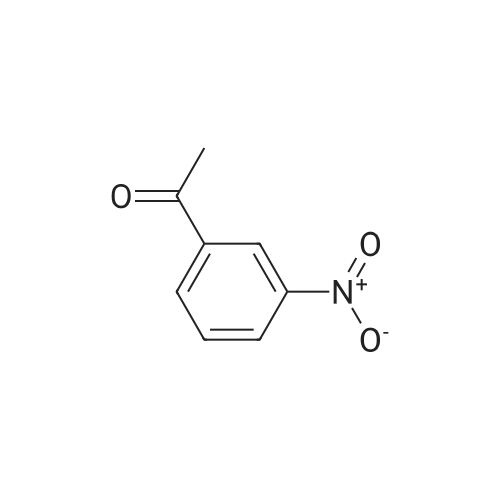
1-(3-Nitrophenyl)ethanol (ID30) To a solution of 1-(3-nitrophenyl)ethanone (ID29, 2.32 g, 14.0 mmol) in MeOH (100 mL) at 0 C. was added NaBH4 (2.20 g, 58.2 mmol) and the resulting mixture was allowed to stir at 0 C. for 1 h. After concentration, the residue was diluted with EtOAc (150 mL) and water (50 mL) and the organic layer was washed with water (50 mL), brine (50 mL), and dried over Na2SO4. After filtration, the filtrate was concentrated and the residue was dried in vacuo to give compound ID30 as a yellowish oil (2.23 g, 95%). 1H NMR (CDCl3, 600 MHz) delta 8.25 (s, 1H), 8.12 (ddd, J=8.4, 1.2, 1.2 Hz, 1H), 7.72 (d, J=7.2 Hz, 1H), 7.52 (dd, J=8.1, 8.1 Hz, 1H), 5.02 (q, J=6.6 Hz, 1H), 2.07 (s, 1H), 1.54 (d, J=6.6 Hz, 3H). |
| 92% |
With 2BH4(1-)*Zn(2+)*Cl2Na2; In acetonitrile; at 20℃; for 0.666667h; |
General procedure: In a round-bottomed flask (10 mL), equipped with a magneticstirrer bar, a solution of acetophenone (0.121 g, l mmol) was prepared in CH3CN(3 mL). To this solution, Zn(BH4)2/2NaCl (0.210 g,1 mmol) was added. The resulting mixture was stirred at room temperature for 60 min. The reaction was monitored by TLC(eluent; Hexane/EtOAc: 10/1). After completion of the reaction, distilled water (5 mL) was added to the reaction mixture and stirred for 5 min. The mixture was extracted with CH2Cl2 (3 ×8 mL) and dried over anhydrous Na2SO4. Evaporation of the solvent followed column chromatography of the resulting crude material over silica gel (eluent; Hexane/EtOAc: 10/1) afforded crystals of 1-phenylethanol (0.l1 g, 93 % yield,Table 2, entry 11). |
| 90% |
With sodium borohydrid; In ethanol; |
EXAMPLE 3 1-(m-Nitrophenyl)ethyl monate A A solution of sodium borohydride (0.4 g, 10 mmol) in ethanol (10 ml) was treated with m-nitroacetophenone (3.3 g, 20 mmol) at 20 C. for 20 mins. The solution was the diluted with aqueous potassium carbonate and extracted with chloroform. The extracts were dried (magnesium sulphate) and evaporated in vacuo to give 1-(m-nitrophenyl)ethanol as an oil (2.9 g, 90%); |
|
With sodium tetrahydroborate; In ethanol; |
a) Synthesis of alpha-methl-3-nitrobenzenemethanol [R1=CH3, X1=X2=H in formula (XVI)] 33 g (199.8 mmol) of 1-(3-nitrophenyl)ethanone [R1=CH3, X1=X2=H in formula (XV)] was weighed and charged into a 500 ml of eggplant type flask, followed by the addition of 120 ml of ethanol. After cooling on an ice water bath, 4.2 g (111 mmol) of NaBH4 was added in portions and the mixture was reacted for 30 minutes under ice cooling bath and for 2 hours at room temperature. The resulting reaction liquid was concentrated by evaporator and the residue was washed with water to quantitatively obtain light brown oil of alpha-methyl-3-nitrobenzene methanol [R1=CH3, X1=X2=H in formula (XVI)]. |
|
With methanol; sodium tetrahydroborate;Cooling with ice; |
Preparation of 2-(4-methylpiperazin-1-yl)ethyl {3-[1-(5,6-difluoro-2-oxobenzoxazol-3-yl)ethyl]phenyl}carbamate (?A27?)Step aPreparation of 1-(3-nitrophenyl)ethanol26.4 g (160 mmol) of 1-(3-nitrophenyl)ethanone are suspended in 270 ml of methanol, and 6.1 g (160 mmol) of sodium borohydride are added in portions with ice cooling. The reaction mixture is subsequently stirred for a further 3 h without cooling, diluted with 300 ml of dichloromethane and washed with 3×200 ml of water. The organic phase is dried over sodium sulfate and evaporated to dryness.Product: 26.15 g; HPLC: Rt=3.87 min (method A). |
|
|
General procedure: All racemic alcohols except for the (R,S)-1-phenylethanol were prepared using the following procedure: a mixture of ketone (5 mmol) and sodium borohydride (10 mmol) in anhydrous methanol (50 mL) was stirred at room temperature for 30 min. Next, saturated NaHCO3 (50 mL) and CH2Cl2 (100 mL) were introduced, and the mixture was stirred at room temperature for another 10 min. The organic layer was removed, and the aqueous layer was extracted twice with 25 mL CH2Cl2. The combined organic layers were dried over anhydrous Na2SO4 and concentrated to give the corresponding crude alcohols. The crude alcohols were then chromatographed to afford pure products. The 1H NMR spectra of these products were in good agreement with the literature. |
| 67%Chromat. |
With sodium hydroxide; In isopropyl alcohol; at 82℃; for 0.75h; |
General procedure: In a typical procedure, a 5 mg (0.77 mol%) of RuO2/MWCNT and 80 mg (2 mmol) of NaOH were stirred with 5 mL of i-PrOH taken in an ace pressure tube equipped with a stirring bar. Then the substrate (1 mmol) was added to the stirring solution and then the mixture was heated at 82C. The completion of the reaction was monitored by GC. After the reaction, the catalyst was separated out from the reaction mixture by simple centrifugation and the products and unconverted reactants were analyzed by GC without any purification. Selectivity of the product for each reaction was alsocalculated. Finally, the separated RuO2/MWCNT was washed well with diethyl ether followed by drying in an oven at 60C for 5 h and it was reused for the subsequent transfer hydrogenation of carbonyl compounds to investigate the reusability of the RuO2/MWCNT. |
|
With sodium tetrahydroborate; In methanol; at 0℃; for 4h; |
General procedure: To a solution of the corresponding ketone in methanol (0.60 M) was added sodiumborohydride (0.33 equiv.) and the resulting mixture was stirred for 2 h at 0 C. Afterstirring, additional sodium borohydride (0.33 equiv.) was added to the mixture tocomplete the transformation. After 2 h, water was added to quench the reaction, andthe mixture was extracted with diethyl ether (3 times). The extracts were dried overanhydrous MgSO4, filtered, and concentrated under reduced pressure. The residuewas chromatographed on silica gel to afford desired alcohol. |
|
With methanol; sodium tetrahydroborate; at 0 - 20℃; |
General procedure: Ten mmol of NaBH4 was added to a cooled (0C) solution of 2.5 mmol of each specific substrate(1a-1f, 1m and 1n) in 50 mL of methanol. After stirring for 10 min, the mixture was warmed to room temperature and stirred for another 3-4 h to complete the reduction. After quenching with 2 M HCl topH 7.0, the mixture was extracted with EtOAc (50 mL 3). The organic phases were washed withbrine, dried over Na2SO4, filtered and concentrated in vacuum. The residue was purified by flashchromatography on silica gel (eluent: EtOAc/PE 1:20) to give the racemic alcohol 2a-2f, 2m and 2n(see Supplementary Materials for NMR spectroscopic data). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping