|
|
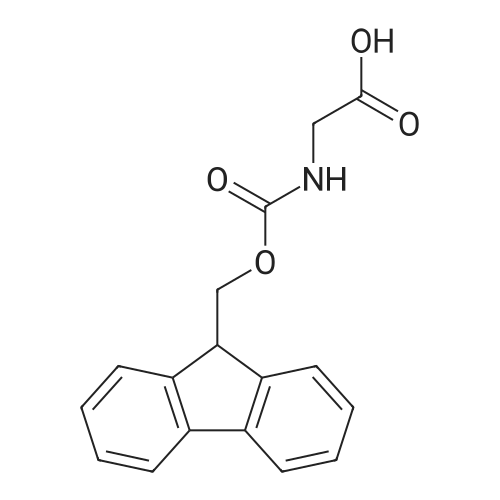
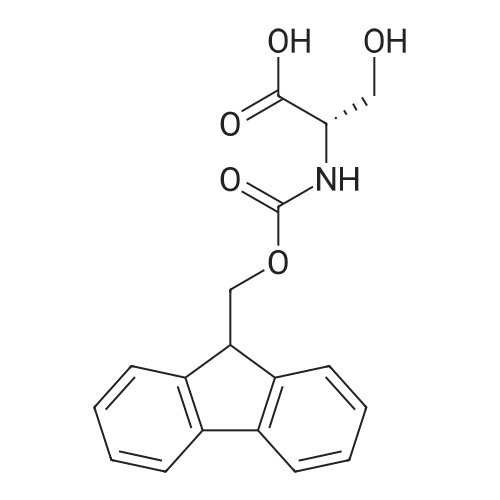
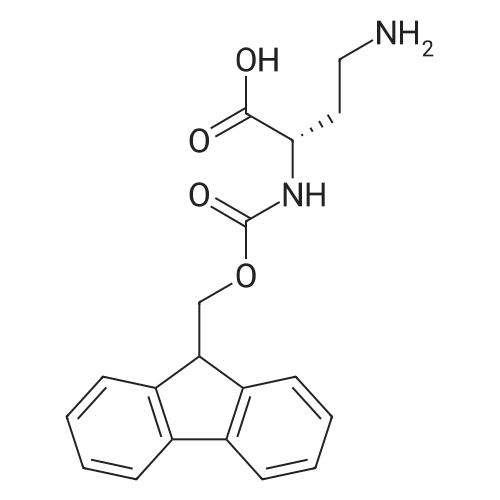
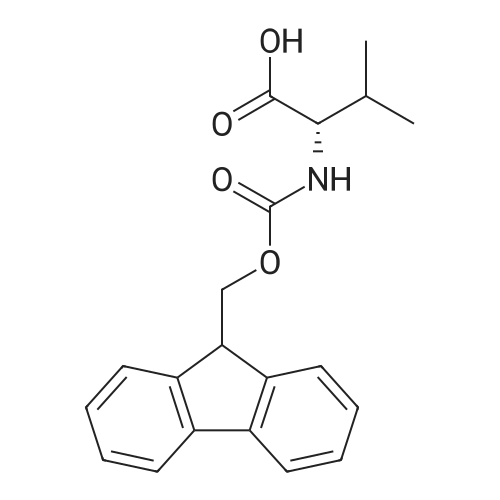
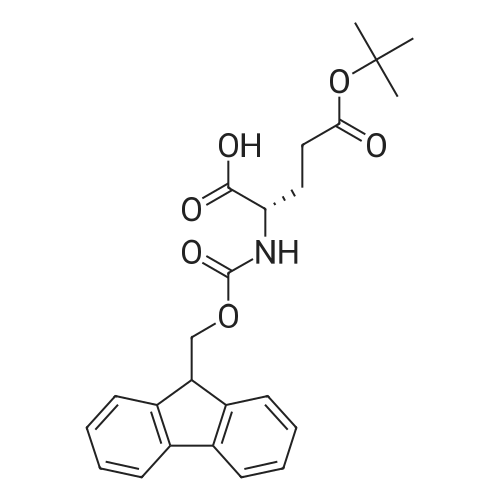
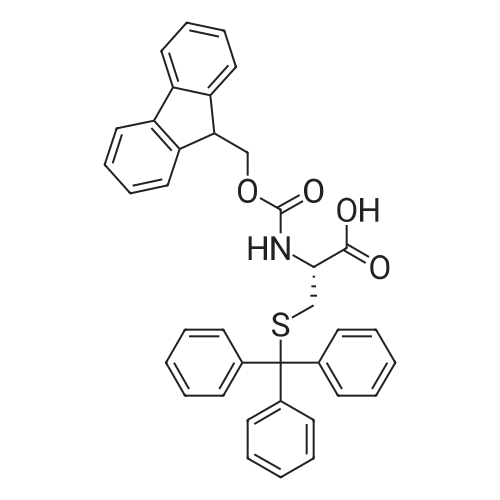
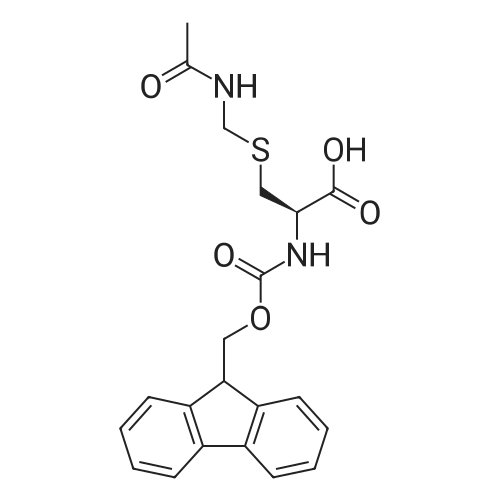
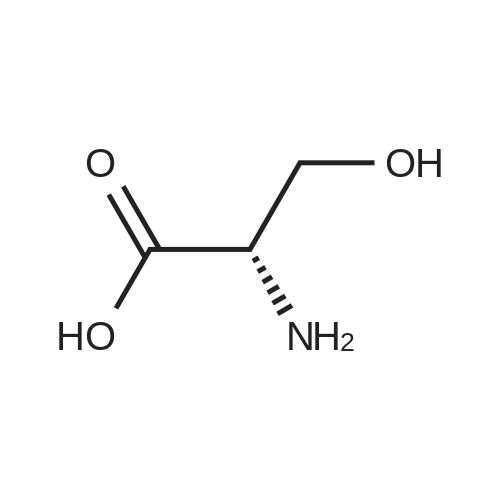
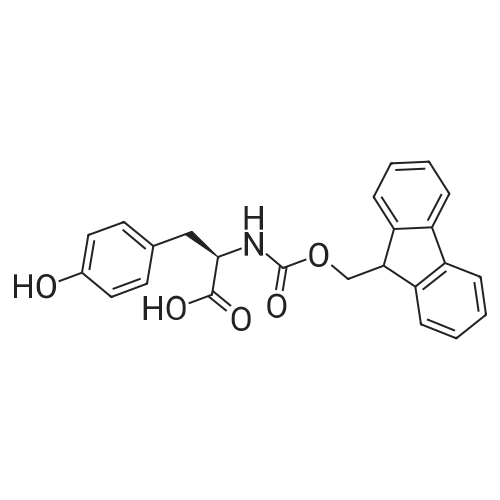
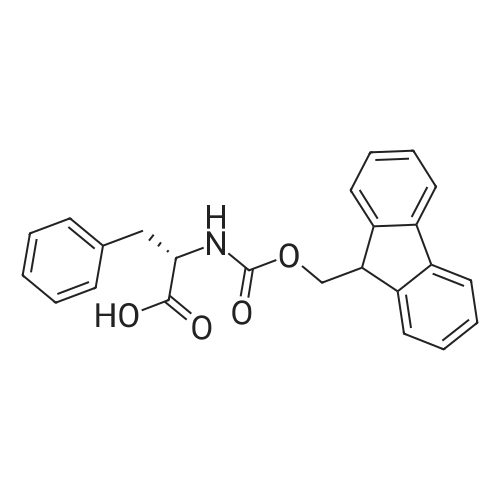
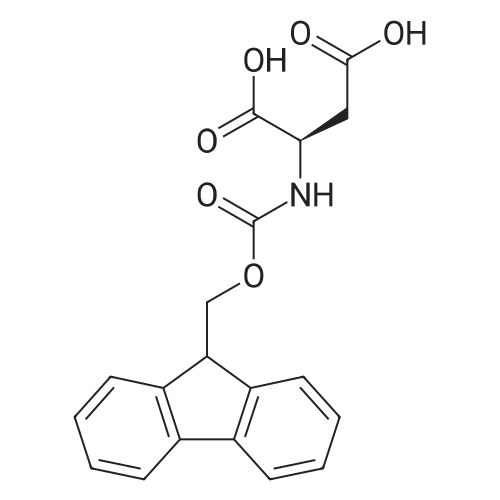
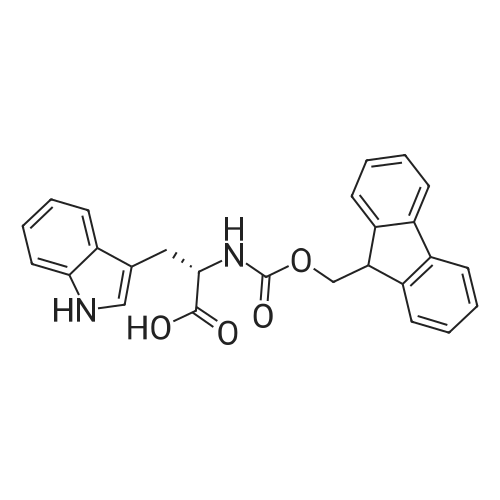
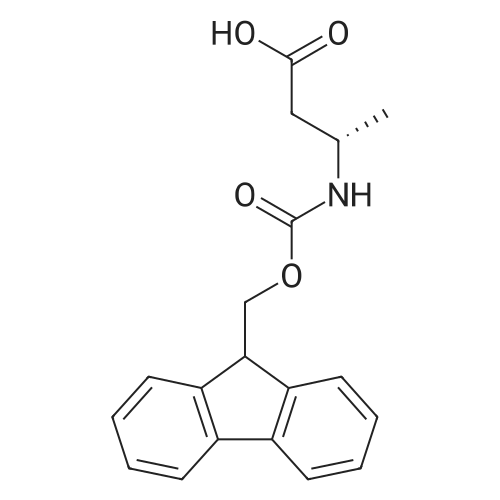
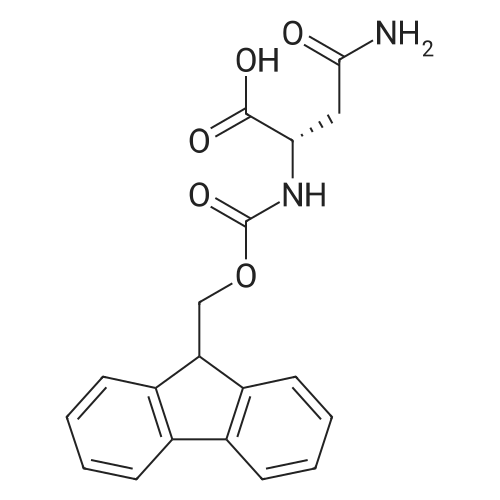
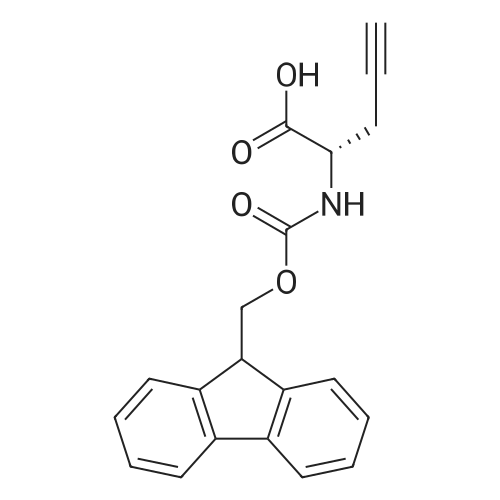
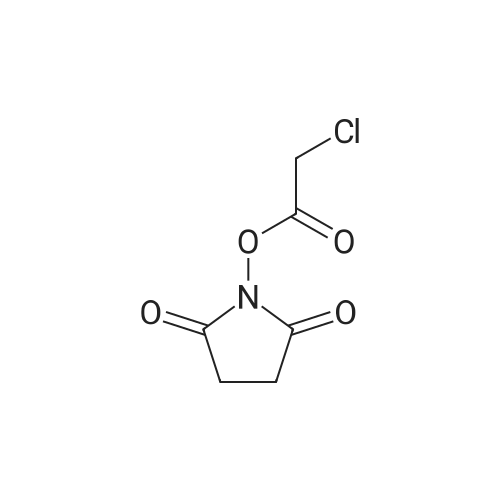
Coupling of the First Protected Amino Acid Residue to the Resin 0.5 g of 2-chlorotritylchloride resin (100-200 mesh, copoly(styrene-1% DVB) polymer matrix, Cat. No. 01-64-0114, Novabiochem, Merck Biosciences Ltd.) (Barlos et al. Tetrahedron Lett. 1989, 30, 3943-3946) (1.4 mMol/g, 0.7 mmol) was filled into a dried flask. The resin was suspended in CH2Cl2 (2.5 ml) and, allowed to swell at room temperature under constant stirring for 30 min. The resin was treated with 0.49 mMol (0.7 eq) of the first suitably protected amino acid residue and 488 mul (4 eq) of diisopropylethylamine (DIEA) in CH2Cl2 (2.5 ml), the mixture was shaken at 25 C. for 4 hours. The resin was shaken (CH2Cl2/MeOH/DIEA: 17/2/1), 30 ml for 30 min; then washed in the following order with CH2Cl2 (1×), DMF (1×), CH2Cl2 (1×), MeOH (1×), CH2Cl2 (1×), MeOH (1×), CH2Cl2 (2×), Et2O (2×) and dried under vacuum for 6 hours. Loading was typically 0.6-0.9 mMol/g. The following preloaded resin was prepared: Fmoc-Pro-2-chlorotritylresin. Synthesis of the Fully Protected Peptide Fragment The synthesis was carried out on a Syro-peptide synthesizer (MultiSynTech GmbH) using 24 to 96 reaction vessels. In each vessel were placed approximately 60 mg (weight of the resin before loading) of the above resin. The following reaction cycles were programmed and carried out: Steps 3 to 6 are repeated to add each amino-acid. Analytical Method: Analytical HPLC retention times (RT, in minutes) were determined using a Jupiter Proteo 90 A column, 150×2.0 mm, (cod. 00E-4396-B0-Phenomenex) with the following solvents A (H2O+0.1% TFA) and B (CH3CN+0.1% TFA) and the gradient: 0 min: 95% A, 5% B; 0.5 min: 95% A, 5% B; 20 min: 40% A, 60% B; 21 min: 0% A, 100% B; 23 min: 0% A, 100% B; 23.1 min: 95% A, 5% B; 31 min: 95% A, 5% B. Formation of Disulfide beta-Strand Linkage After formation of the disulfide beta-strand linkage, the resin was suspended in 1 ml (0.14 mMol) of 1% TFA in CH2Cl2 (v/v) for 3 minutes and filtered, and the filtrate was neutralized with 1 ml (1.15 mMol) of 20% DIEA in CH2Cl2 (v/v). This procedure was repeated twice to ensure completion of the cleavage. The resin was washed three times with 1 ml of CH2Cl2. The CH2Cl2 layer was evaporated to dryness. The volatiles were removed and 8 ml dry DMF were added to the tube. Then 2 eq. of HATU in dry DMF (1 ml) and 4 eq. of DIPEA in dry DMF (1 ml) were added to the peptide, followed by stirring for 16 h. The volatiles were evaporated to dryness. The crude cyclised peptide was dissolved in 7 ml of CH2Cl2 and extracted with 10% acetonitrile in H2O (4.5 ml) three times. The CH2Cl2 layer was evaporated to dryness. To deprotect the peptide fully, 3 ml of cleavage cocktail TFA:TIS:H2O (95:2.5:2.5) were added, and the mixture was kept for 2.5 h. The volatiles were evaporated to dryness and the crude peptide was dissolved in 20% AcOH in water (7 ml) and extracted with isopropyl ether (4 ml) for three times. The aqueous layer was collected and evaporated to dryness, and the residue was purified by preparative reverse phase HPLC. After lyophilisation the products were obtained as white powders and analysed by the HPLC-ESI-MS analytical method described above. The analytical data comprising purity after preparative HPLC and ESI-MS are given. The peptide was synthesized starting with the amino acid L-Pro which was grafted to the resin. Starting resin was Fmoc-Pro-2-chlorotrityl resin, which was prepared as described above. The linear peptide was synthesized on solid support according to the procedure described above in the following sequence: Resin-Pro-DPro-Lys-Gln-Tyr-Cys-Tyr-Arg-Dab-DPro-Ala-Ser-Cys-Ala-His-Tyr. A disulfide beta-strand linkage was introduced as described above. The product was cleaved from the resin, cyclized, deprotected and purified as indicated by preparative reverse phase LC-MS. After lyophilisation the product was obtained as white powder and analysed by the HPLC-ESI-MS analytical method described above ([M+2H]2+: 933.1; RT: 10.47; UV-purity: 72%). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping