There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
Structure of 109-01-3 * Storage: {[proInfo.prStorage]}
1-Methylpiperazine (CAS: 109-01-3) can be used in the preparation of Bosutinib (SKI-606) (CAS: 380843-75-4). Bosutinib, a small molecule that inhibits BCR-ABL and src tyrosine kinases, is utilized for treating chronic myelogenous leukemia.
Application In Synthesis of [ 109-01-3 ]
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With sodium hydroxide In acetone at 20℃; for 24 h; Cooling with ice
N-methylpiperazine (30 mmol, 3.3 ml), 4 ml of 25percent NaOH solution and 40 ml of acetone were added to a 100 ml round-bottomed flask under ice-cooling, then 1-bromo-3-chloropropane (30 mmol, 3 ml) After completion of ice bath stirring to insoluble material dissolved, and then at room temperature for 24h.The solvent was concentrated under reduced pressure, 20 ml of water-soluble concentrate was added, extracted with methylene chloride, dried over anhydrous sodium sulfate, filtered and concentrated to give a clear oil. The reaction flask was placed in an ice bath and 50 ml of ethyl acetate was added. 2.5 ml of concentrated hydrochloric acid was added dropwise slowly until a large amount of white solid was formed. The pH was controlled to about 2 during the reaction. The solvent was concentrated under reduced pressure, recrystallized from 100 ml of anhydrous ethanol, filtered and dried to obtain 3.3 g of a white solid,
25%
Stage #1: With sodium hydroxide In water; acetone at 0 - 20℃; for 24 h; Stage #2: With hydrogenchloride In ethanol
A modified procedure of Mahesh et al, Pharmazie, 2005, 60, 6, 411-414, was used. After cooling a stirred solution of N-methylpiperazine (50 mmol, 5.55 ml) in 100 ml acetone to 0 °C, 10 ml of an aqueous 25 percent NaOH-solution and l-bromo-3-chloropropane (50 mmol, 7.87 g = 4.92 ml) were added cautiously. The reaction was stirred at RT for 24 hours. After concentrating the mixture under reduced pressure, the residue was diluted with water and extracted with dichloromethane. The collected organic phases were dried over Na2S04, filtered and concentrated. The residue was diluted with ethanol and after adding 2.3 M ethanolic HC1 l-(3-chloropropyl)-4-methylpiperazin-dihydrochloride crystallized as white crystals(12.5 mmol, 25 percent). Mp = 257 °C. ]H NMR (300 MHz, DMSO) 3.74 (t; 2H; 3J = 6.4 Hz; NCH7CH7CH7CI); 3.37 (m; 12H: NCH7CH7CH7Cl+4xpiperazin-CH7+2x H); 2.81 (s; 3H; CH3); 2.19 (d; 2H; J = 6.8 Hz; NCH2CH2CH2CI).
Reference:
[1] Patent: CN105884699, 2016, A, . Location in patent: Paragraph 0044; 0045
[2] Patent: WO2011/73092, 2011, A1, . Location in patent: Page/Page column 29; 30
2
[ 109-01-3 ]
[ 555-16-8 ]
[ 70261-81-3 ]
Yield
Reaction Conditions
Operation in experiment
90%
With sodium tetrahydroborate; acetic acid In chloroform at 0 - 20℃; for 13 h;
General procedure: AcOH (100percent) (140 mL, 2.44 ml) was added over 1 h to a flask containing stirred NaBH4 (20.0 g, 0.53 ml) and CHCl3 (220 mL) at 0-5 °. The resulting mixture was stirred at 0-5 ° for 1.5 h and 1-methylpiperazine (1) (28.0 ml, 0.25 ml) and a solution of methyl 4-formylbenzoate (2a) (43.4 g, 0.26 ml) in CHCl3 (60 mL) were added. The resulting mixture was stirred at 0-5 ° for 1 h and then for 12 h at rt. the mixture was treated with H2O (150 mL) and Na2CO3 until pH 8.0-9.0. The aqueous phase was extracted with EtOAc (2 .x. 100 ml) then both organic layers were combined, washed with H2O (1 .x. 100 ml), and dried over anhydrous Na2SO4. Filtration and evaporation of the solvents gave methyl 4-[(4-methylpiperazin-1-yl)methyl]benzoate (4a): yellowish oil; yield: 61.6 g, 99percent.
Reference:
[1] Tetrahedron Letters, 2012, vol. 53, # 38, p. 5056 - 5058
[2] Russian Journal of Organic Chemistry, 2013, vol. 49, # 4, p. 563 - 567[3] Zh. Org. Khim., 2013, vol. 49, # 4, p. 580 - 584
[4] Journal of Medicinal Chemistry, 2017, vol. 60, # 21, p. 8801 - 8815
[5] Russian Journal of Organic Chemistry, 2011, vol. 47, # 10, p. 1556 - 1563
3
[ 109-01-3 ]
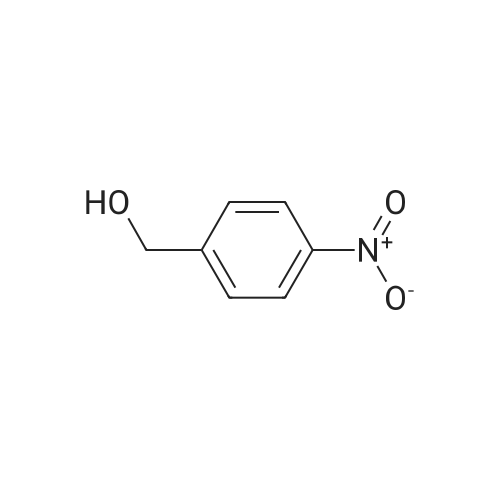
[ 100-11-8 ]
[ 70261-81-3 ]
Yield
Reaction Conditions
Operation in experiment
92%
With potassium carbonate; potassium iodide In acetonitrile for 5 h;
4-nitrobenzylbromide (4.30 g, 20 mmol) was dissolved in 40 ml of acetonitrile solvent,Then K2CO3 (5.52 g, 40 mmol) was added,KI (0.33 g, 2 mmol),Then, 4-methylpiperazine (2.20 g, 22 mmol) was added thereto,After 5 h, the reaction was complete.The reaction solution was concentrated and dried,And further adding 100 ml of H2O thereto,Extracted with ethyl acetate (150 ml x 3)The organic layer was collected, concentrated,4.32 g of a solid was obtained in 92percent yield.
Reference:
[1] Medicinal Chemistry Research, 1999, vol. 9, # 3, p. 149 - 161
[2] Patent: CN106432235, 2017, A, . Location in patent: Paragraph 0220; 0221
[3] Bioorganic and Medicinal Chemistry, 2010, vol. 18, # 15, p. 5738 - 5748
[4] Patent: WO2005/54238, 2005, A1, . Location in patent: Page/Page column 88
[5] Chemical and Pharmaceutical Bulletin, 2014, vol. 62, # 3, p. 238 - 246
[6] Patent: EP2955185, 2015, A1, . Location in patent: Paragraph 0079
[7] Patent: CN103739550, 2016, B, . Location in patent: Paragraph 0173-0176
[8] European Journal of Medicinal Chemistry, 2017, vol. 130, p. 86 - 106
[9] Patent: CN107245073, 2017, A, . Location in patent: Paragraph 0121; 0122; 0123
[10] Journal of Medicinal Chemistry, 2017, vol. 60, # 21, p. 8801 - 8815
[11] Journal of Medicinal Chemistry, 2018, vol. 61, # 4, p. 1499 - 1518
[12] European Journal of Medicinal Chemistry, 2018, vol. 155, p. 303 - 315
4
[ 109-01-3 ]
[ 100-14-1 ]
[ 70261-81-3 ]
Yield
Reaction Conditions
Operation in experiment
50%
at 20℃; for 24 h;
The First Step: Preparation of 1-methyl-4-(4-nitrobenzyl)-piperazine (F652-01) Monomethylpiperazine (15 mL) and tetrahydrofuran (60 mL) were placed in a 200 mL eggplant shaped flask, and a solution of 4-nitrobenzylchloride (8.58 g, 50 mmol) in tetrahydrofuran was added dropwise to the mixture at room temperature while stirring. After finishing the instillation, the mixture was stirred at room temperature for 24 hours. The reaction mixture was mixed with distilled water, and the precipitated solids were collected by filtration and dried under reduced pressure to obtain the title compound (5.9 g, 50percent). LC/MS (Method 3): m/z (ESI, POS): 236[M+H]+; retention time: 1.28 minutes.
59%
With sodium carbonate; triethylamine In ethylene glycol
A. 1-methyl-4-[(4-nitrophenyl)methyl]piperazine To a solution of p-nitrobenzyl chloride (5.2 g, 10mM) and 3.2 g of triethylamine in 30 ml of ethylene glycol is added a solution of N-methylpiperazine (3 g, 30 mM) in 20 ml of ethylene glycol. After complete addition, the resulting solution is heated to 80° C. under nitrogen for 30 minutes. The reaction mixture is quenched into aqueous 10percent sodium carbonate solution and extracted with methylene chloride. The methylene chloride solution is washed with water, saturated sodium chloride solution, dried over anhydrous sodium sulfate and concentrated to give 4.15 g (59percent) of 1-methyl-4-[(4-nitrophenyl)methyl]piperazine.
Reference:
[1] Farmaco, 1992, vol. 47, # 3, p. 335 - 344
[2] Patent: EP1857446, 2007, A1, . Location in patent: Page/Page column 45
[3] Patent: WO2008/94575, 2008, A2, . Location in patent: Page/Page column 52-53
[4] European Journal of Medicinal Chemistry, 2011, vol. 46, # 7, p. 2917 - 2929
[5] Patent: US4140775, 1979, A,
31.5 g of 4-chloro-1-nitrobenzene and 44.4 ml of 1-methyl-piperazine are combined and stirred for 18 hours at 90° C. Then the solution is poured onto ice water and the precipitate formed is suction filtered, washed with water and recrystallised from ethanol/water (1:1). The residue is dried in vacuo at 75° C. [00369] Yield: 44.0 g (99percent of theory), [00370] Rf value: 0.5 (silica gel, methylene chloride/methanol=10:1) [00371] Melting point: 108-112° C.
Reference:
[1] Chemistry of Heterocyclic Compounds, 2007, vol. 43, # 12, p. 1540 - 1543
8
[ 109-01-3 ]
[ 100986-89-8 ]
[ 138199-71-0 ]
Reference:
[1] Patent: US2003/130507, 2003, A1,
9
[ 109-01-3 ]
[ 342417-00-9 ]
[ 342417-01-0 ]
Yield
Reaction Conditions
Operation in experiment
95%
at 100℃; for 2 h;
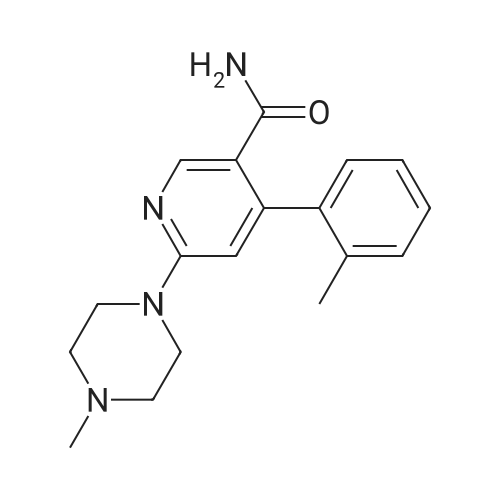
Step 3 1.0 g (4.05 mMol) 6-Chloro-4-o-tolyl-nicotinamide in 9.0 ml 1-methyl-piperazine was heated to 100° C. for 2 hours. The excess N-methyl-piperazine was removed under high vacuum and the residue was filtered on silica gel (eluent: dichloromethane) to yield 1.2 g (95percent) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide as a light yellow crystalline foam. MS (ISP): 311 (M+H+, 100), 254 (62).
95%
at 100℃; for 2 h;
Step 3: (0177) 1.0 g (4.05 mMol) 6-Chloro-4-o-tolyl-nicotinamide in 9.0 ml 1-methyl-piperazine was heated to 100° C. for 2 hours. The excess N-methyl-piperazine was removed under high vacuum and the residue was filtered on silica gel (eluent: dichloromethane) to yield 1.2 g (95percent) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide as a light yellow crystalline foam. (0178) MS (ISP): 311 (M+H+, 100), 254 (62).
Reference:
[1] Patent: US8426450, 2013, B1, . Location in patent: Page/Page column 25
[2] Patent: US9403772, 2016, B2, . Location in patent: Page/Page column 30
[3] Journal of Organic Chemistry, 2006, vol. 71, # 5, p. 2000 - 2008
[4] Patent: JP2015/17121, 2015, A, . Location in patent: Paragraph 0153
10
[ 109-01-3 ]
[ 342417-01-0 ]
Reference:
[1] Organic Process Research and Development, 2006, vol. 10, # 6, p. 1157 - 1166
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In pyridine;
EXAMPLE L 4-(4-Methylpiperazinyl)-1,8-naphthalic anhydride 4-Methylpiperazine (0.6 g, 6.6 mmol) and DBU (1 mL) were added to <strong>[21563-29-1]4-bromo-1,8-naphthalic anhydride</strong> (1.5 g, 5.4 mmol) in pyridine (10 mL). The solution was refluxed for 8 hours, concentrated in vacuo, and the residue was triturated with water. The separated solid was filtered and dried to give 0.8 g of the title compound.
8-chloro-2-[3-(4-phenyl-3,6-dihydro-1 (2H)-pyridinyl)propyl]-4(3H)-quinazolinone[ No CAS ]
[ 3375-31-3 ]
[ 224311-51-7 ]
[ 437999-29-6 ]
Yield
Reaction Conditions
Operation in experiment
In tetrahydrofuran; methanol; dichloromethane; toluene;
EXAMPLE 15 A mixture of 8-chloro-2-[3-(4-phenyl-3,6-dihydro-1 (2H)-pyridinyl)propyl]-4(3H)-quinazolinone (50 mg), 1-methylpiperazine (19.8 mg), palladium (II) acetate (2.96 mg), 2-(di-t-butylphosphino)biphenyl (7.86 mg), sodium t-butoxide (23 mg) in toluene (0.4 ml and tetrahydrofuran (0.2 ml) was stirred at 80° C. under nitrogen atmosphere overnight. The mixture was cooled, diluted with water and extracted with dichloromethane twice. The combined extracts were dried over magnesium sulfate and concentrated. The residue was purified by preparative thin layer chromatography on silica gel using 10percent methanol in dichloromethane to give the 8-(4-methyl-1-piperazinyl)-2-[3-(4-phenyl-3,6-dihydro-1(2H)-pyridinyl)propyl]-4(3H)-quinazolinone. Mass (APCI): 444.3 (M++H)
(E)-ethyl α-cyano-2-(4-methylpiperazine)-5-cyanocinnamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
52%
In ethanol; at 78℃; for 1.75h;
General procedure: Cyclic secondary amine (2.5 equiv) was treated with 2-halobenzaldehyde (1 mmol) and active methylidene compound (1 mmol) in EtOH (2 mL) or DMF (2 mL). The mixture was stirred and heated to reflux. After the reaction was completed, the mixture was cooled down to r.t. and poured into H2O (10 mL). Crude products were filtered off and purified by recrystallization in EtOH or by column chromatography on silica gel; 52?88percent yield.
With copper(l) iodide; sodium acetate; N-ethyl-N,N-diisopropylamine; In 1,4-dioxane; at 100℃; for 4h;Inert atmosphere;
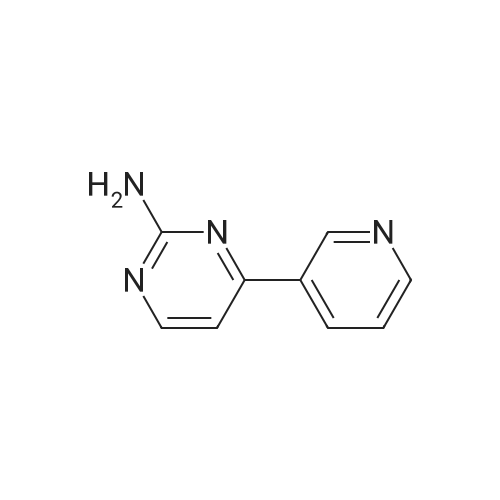
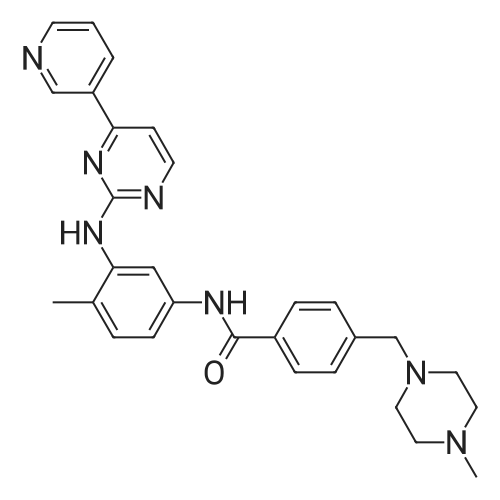
Under nitrogen, 10 mmol of 4- (3-pyridyl) -2-aminopyrimidine (CAS: 66521-66-2)10 mmol of 4-chloromethylbenzoyl (3-bromo-4-methylphenyl) amine (CAS: 1072105-05-5), 10 mmol of N-methylpiperazine (CAS: 109-01-3 ) And 0.2 mmol of cuprous iodide,0.2 mmol of N, N'-diisopropylethylenediamine,110 mmol of sodium acetate in 20 mL of 1,4-dioxane,The reaction was carried out under heating at a temperature of 100 ° C for 4 hours.After completion of the reaction, the solvent was evaporated to dryness with a rotary evaporator and the residue was separated by column chromatography,Obtain 4.2 grams of imatinib in 86percent yield.
With cesiumhydroxide monohydrate; In dimethyl sulfoxide; at 120℃; for 20h;
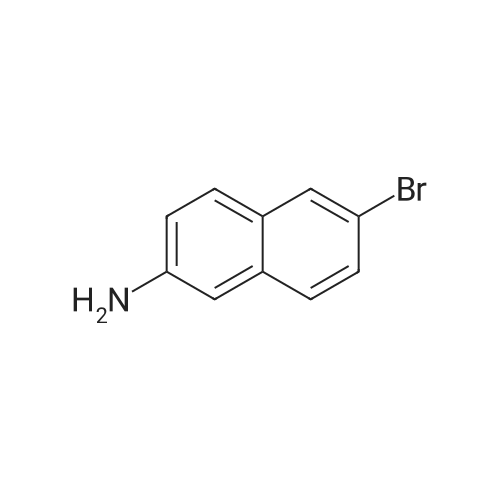
<strong>[7499-66-3]6-bromo-2-aminonaphthalene</strong> (500 mg, 2.25 mmol),N-methylpiperazine (270 mg, 2.7 mmol),cesium hydroxide hydrate(760 mg, 4.5 mmol)A solution of dimethyl sulfoxide (5.0 mL) was stirred at 120 ° C for 20 hours.The reaction system was then cooled to room temperature.Dilute with ice water (10 mL).The aqueous phase was extracted with dichloromethane (20 mL×2).Organic phase in turn,Washed with saturated saline,Dry over anhydrous sodium sulfate,filter,concentrate,The residue was purified by flash column chromatography ( petroleum ether / ethyl acetate = 1/3)Compound 21.1 (70 mg, yield: 13percent) was obtained as a brown solid.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +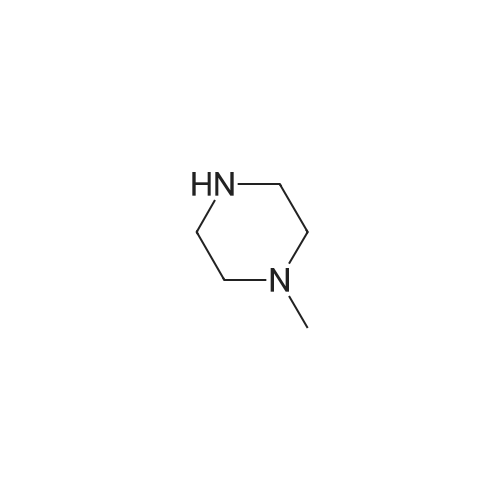

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping