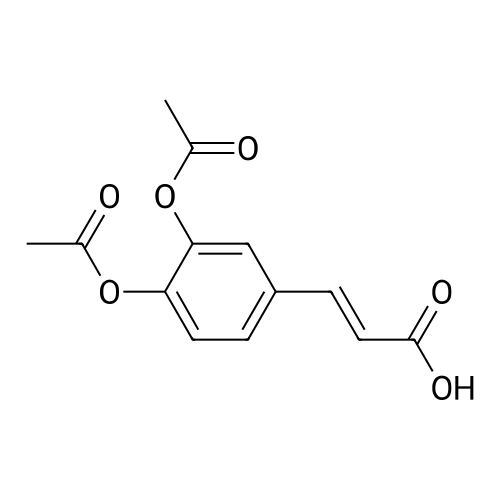
| 48% |
With ytterbium(III) triflate; In nitromethane; at 120℃; for 0.666667h;Catalytic behavior; |
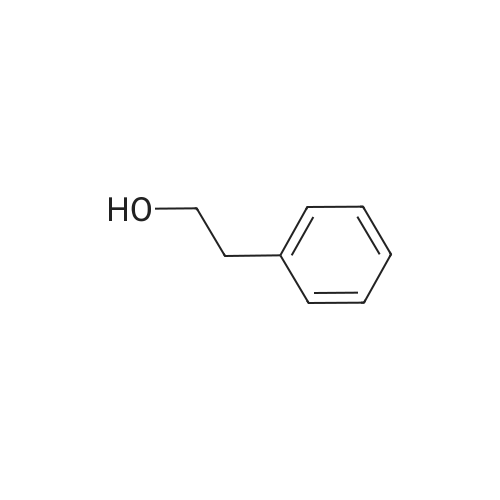
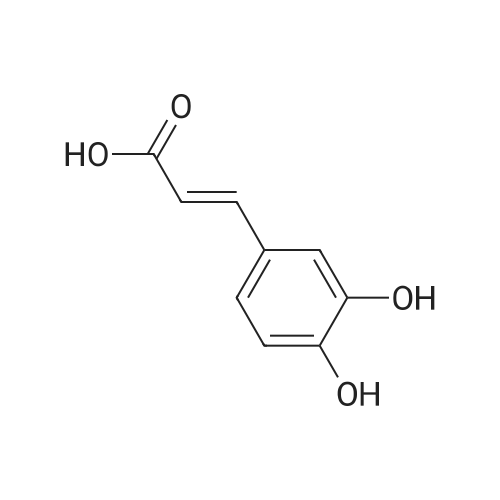
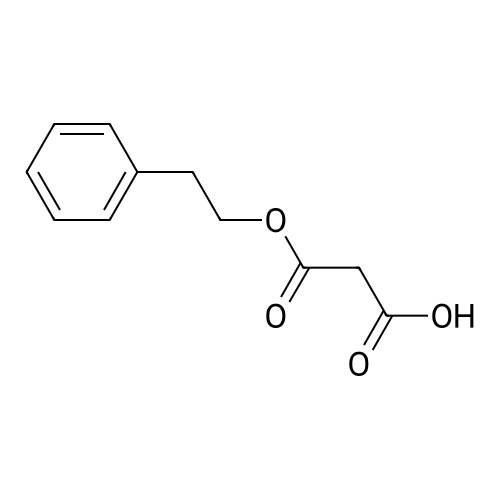
To a mixture of caffeic acid fine powder (1.0 g, 5.56 mmol, 1.0 equiv.),alcohol (5.56 mmol, 1.0 equiv.) in nitromethane (125 mL) was addedytterbium triflate (34.4 mg, 0.056 mmol, 0.01 equiv.). After 5 min inan ultrasonic bath the mixture without protective gas was stirred ona 120 C oil bath for a given time. The reaction mixture was cooled toroom temperature, washed with deionised water (30 mL), 2% NaHCO3(30 mL) and brine, dried over anhydrous Na2SO4 and evaporated underreduced pressure to give the crude product, which was purified on asilica gel column to give the compounds 1-5 and 8-30.2-Phenethyl (E)-3-(3,4-dihydroxyphenyl) acrylate (1): Whitesolid; yield 758 mg, 48.0%; m.p. 128-130 C (lit.20 116-123 C);IR (KBr) numax 3480, 3328, 1683, 1601, 1362, 1301, 1279, 1182 cm-1;1H NMR (400 MHz, DMSO-d6) deltaH 7.46 (1H, d, J = 16 Hz, CH=CHCO),7.34-7.18 (5H, m, C6H5), 7.05 (1H, s, 2-ArH), 6.99 (1H, d, J = 8.0 Hz,6-ArH), 6.77 (1H, d, J = 8.0 Hz, 5-ArH), 6.24 (1H, d, J = 16 Hz,CH=CHCO), 4.32 (2H, t, J = 6.8 Hz, OCH2), 2.94 (2H, t, J = 6.8 Hz,OCH2CH2) ppm; 13C NMR (100 MHz, DMSO-d6) deltaC 166.4, 148.3,145.4, 145.1, 138.0, 128.8, 128.3, 126.3, 125.4, 121.4, 115.7, 114.7, 113.8,64.3, 34.4 ppm; HRMS-ESI C17H16O4 calcd [M-H]- 283.0970, found283.0966. |
| 48% |
With ytterbium(III) triflate; In nitromethane; at 120℃; |
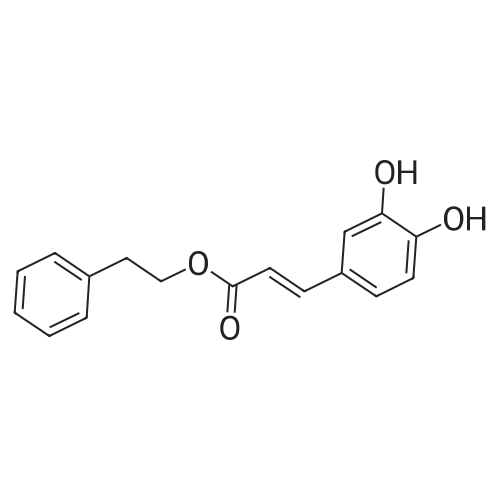
General procedure: To a mixture of caffeic acid fine powder (1.0 g, 5.56 mmol), various phenethyl alcohols (5.56 mmol) in CH3NO2 (125 mL) was added Yb(OTf)3 (34.4 mg, 0.056 mmol). After 5 min of ultrasonic shake, the mixture was stirred on a 120 C oil bath for 40-120 min. The reaction mixture was cooled to room temperature, washed with 2% NaHCO3 (30 mL) and brine, dried over anhydrous Na2SO4, and concentrated to give crude products, which were purified by column chromatography to give the compounds 1-26 in 18-61% yields. Phenethyl (E)-3-(3,4-dihydroxyphenyl) acrylate (CAPE). White solid (48% yield for esterification reaction); Mp 128-130 C; 1H NMR (400 MHz, DMSO-d6) deltaH 7.46 (1H, d, J = 16.0 Hz, CH=CHCO), 7.34-7.18 (5H, m, C6H5), 7.05 (1H, s, 2-ArH), 6.99 (1H, d, J = 8.0 Hz, 5-ArH), 6.77 (1H, d, J = 8.0 Hz, 6-ArH), 6.24 (1H, d, J = 16.0 Hz, CH=CHCO), 4.32 (2H, t, J = 6.8 Hz, OCH2), 2.94 (2H, t, J = 6.8 Hz, CH2C6H5) ppm; 13C NMR (100 MHz, DMSO-d6) deltaC 166.4, 148.3, 145.4, 145.1, 138.0, 128.8, 128.3, 126.3, 125.4, 121.4, 115.7, 114.7, 113.8, 64.3, 34.4 ppm; HRMS-ESI C17H16O4 calcd [M-H]- 283.0970, found 283.0966. |
| 9% |
With ytterbium(III) triflate; In nitromethane; for 1.5h;Reflux; |
Caffeic acid (0.5 g, 2.78 mmol) and 2-phenylethanol (0.35 mL,2.78 mmol) were dissolved in nitromethane (62.5 mL). Yb(OTf)3(0.017 g, 0.028 mmol) was added, and the suspension was stirredfor 5 min in an ultrasonic bath followed by an additional 1.5 hstirring under reflux [4]. The reaction mixture was stirred at roomtemperature overnight and washed with NaHCO3-solution (2%,15 mL) and brine (15 mL). The organic phase was dried over Na2SO4and concentrated under reduced pressure. The crude product waspurified by column chromatography (silica gel; chloroform/methanol,99:1), and compound 17 was obtained as a white solid (0.07 g,9%); RF 0.04 (silica gel; chloroform/methanol, 99:1); m.p.128-130 C (lit.: [19]: 126-128 C); IR (KBr): nu 3480s, 1685m,1636m, 1602s, 1535w, 1442w, 1363w, 1302m, 1279s, 1182s cm1;UV-vis (CHCl3): lambda (log epsilon) 250 (4.06), 320 (4.21), 353 (4.28) nm; 1HNMR (500 MHz, CDCl3): delta 7.56 (d, J 15.9 Hz, 1H, 3-H), 7.34-7.30(m, 2H, 2-H), 7.27-7.22 (m, 3H, 3-H4-H), 7.07 (d, J 2.0 Hz, 1H, 50-H), 7.01 (dd, J 8.2, 2.0 Hz, 1H, 20-H), 6.87 (d, J 8.2 Hz, 1H, 60-H),6.25 (d, J 15.9 Hz, 1H, 2-H), 4.42 (t, J 7.1 Hz, 2H, 1-H), 3.01 (t,J 7.1 Hz, 2H, 2-H); 13C NMR (125 MHz, CDCl3): delta 167.8 (C-1),146.5 (C-40), 145.2 (C-30), 143.9 (C-3), 138.0 (C-1), 129.1 (C-3), 128.7(C-2), 127.7 (C-10), 126.8 (C-4), 122.7 (C-60), 115.7 (C-2), 115.6 (C-50),144.6 (C-20), 65.3 (C-1), 35.4 (C-2) ppm; MS (ESI, MeOH): m/z(%) 285.0 ([MH], 45), 302.2 ([MNH4], 23), 307.1 ([MNa],100), 446.1 ([3 MKH]2, 44), 580.1 ([4 MNaH]2, 23), 588.0([4MKH]2, 50), 590.8 ([2MNa], 42), 599.9 ([4MZn]2, 48);analysis calcd for C17H16O4 (284.31): C 71.82, H 5.67; found: C 71.69,H 5.83. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping