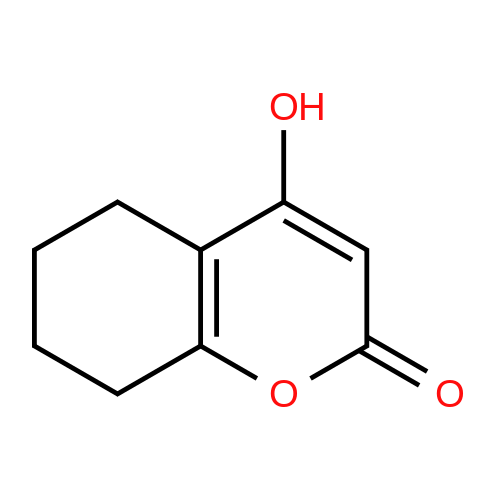
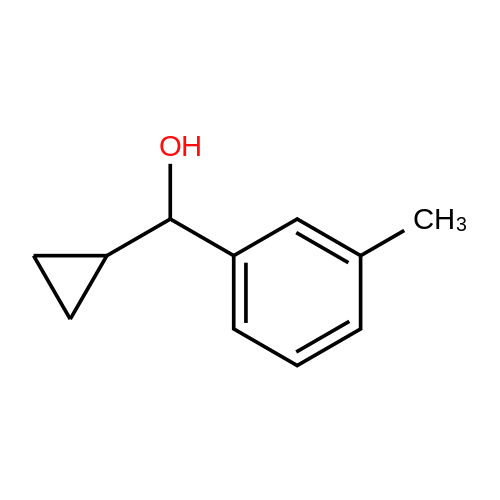
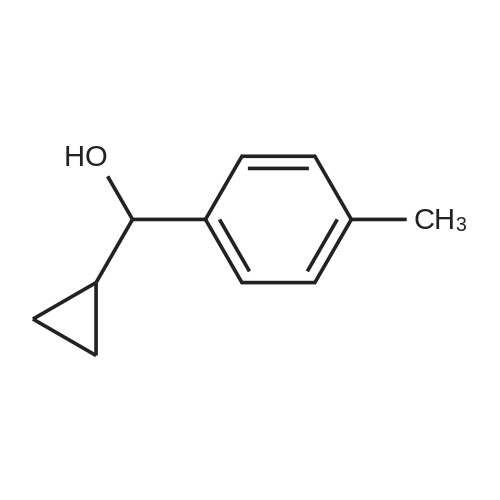
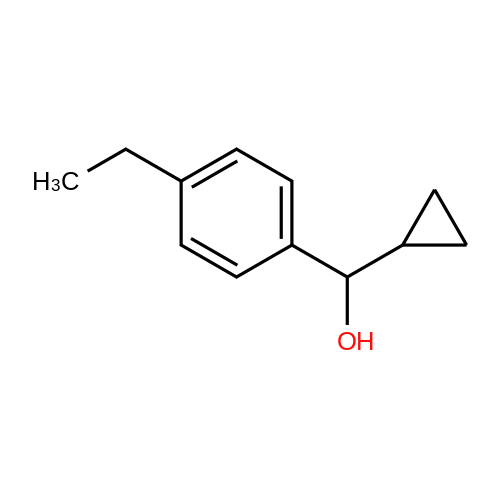
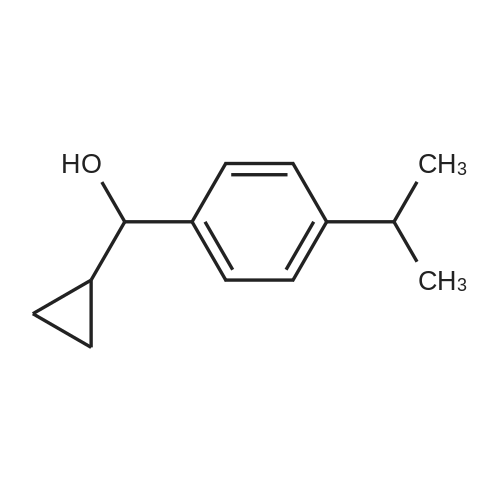
| 80% |
With sodium tetrahydridoborate; In methanol; at 20℃; for 4h; |
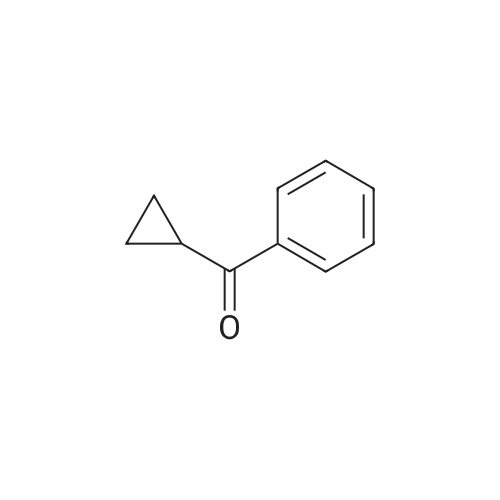
General procedure: For a typical reaction, NaBH4 (1.0g, 26.8mmol) was added to a stirred solution of 1-pentanophenone (4.4g, 26.8mmol) in dry methanol (30mL). The exotherm was controlled by an ice bath. The suspension was stirred at room temperature for 4h, monitored by TLC. After the reaction was quenched by an addition of water (20mL), the methanol was removed under vacuum and the residue was extracted with ethyl acetate (3×30mL). The combined organic phases were washed with brine (20mL), dried over MgSO4 and then filtered. The organic solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography (hexane/ethyl acetate, 3:1) to give rac-4a (3.8g, 83%). The 1H NMR spectra of alcohols 3a,24 4a,24 5a,24 6a,24 7a,25 8a,26 9a,27 were all in agreement with those reported in the literature. |
| 74% |
With 5,11,17,23-tetra-tert-butyl-25,27-dihydroxy-26,28-dipropoxy-calix<4>arene; isopropanol; In hexane; toluene; at 20℃; for 12h; |
General procedure for conducting MPV reduction: In 5 mL of anhydrous toluene (freshly distilled over sodium benzophenone ketyl), 1,3-bis-substituted calix[4]arene (e.g., for 1,3-bispropoxy calix[4]arene, 73 mg, 0.1 mmol, 10 mol%) is dissolved. To this solution, Me3Al (40 μL, 0.1 mmol, 10 mol%, 25% w/w solution in hexane) is added under Ar. Gaseous methane bubbles indicate the formation of methylaluminum calix[4]arene complex, which upon treatment with alcohol forms the active MPV catalyst and evolves methane. A 5 mL solution of alcohol (e.g., 300 μL, 240 mg, 4 mmol, 4 equiv. anhydrous 2-propanol) and substrate ketone (e.g., 155 mg, 1 mmol, 1 equiv. α-chloroacetophenone) in toluene is added. The reaction is subsequently performed at room temperature and is monitored by 1H NMR spectroscopy. |
| 70% |
With manganese(I) pentacarbonyl bromide; potassium-t-butoxide; Ethane-1,2-diamine; In isopropanol; at 80℃; for 3h; |
General procedure: To a solution of acetophenone (58 μL, 0.5 mmol) in 2-propanol (0.5 mL) was added a stock solution of manganese pentacarbonyl bromide (0.5 mL, 0.005 mol·L-1; 2.7 mg, 0.010 mmol, in 2 mL 2-propanol)followed, in this order, by a stock solution of ethylenediamine (0.5 mL,0.005 mol·L-1; 1.0 μL, 0.0125 mmol, in 2.5 mL 2-propanol) and tBuOK (0.5 mL, 0.010 mol·L-1; 2.4 mg, 0.020 mmol, in 2 mL 2-propanol). The reaction mixture was stirred for 3 h at 80 C in an oil bath. The solution was then filtered through a small pad of silica (2 cm in a Pasteur pip-ette). The silica was washed with ethyl acetate. The filtrate was evaporated and the conversion was determined by 1H NMR. The crude residue was then puried by column chromatography (SiO 2 , mixture of petroleum ether/ethyl acetate or dietyl ether as eluent. Enantiomeric excesses were determined by GC analyses performedon GC-2014 (Shimadzu) 2010 apparatus equipped with Supelco beta-DEX 120 column (30 m × 0.25 mm). The determination of the absoluteconguration was done by comparison with (S)-alcohol obtained bykinetic resolution of racemic alcohols with Novozym 435 (CandidaAntarctica Lipase B) and by comparison of the retention times with the literature [32-34]. |
|
With sodium tetrahydridoborate; In methanol; at 20℃; for 12h;Inert atmosphere; |
The cyclopropyl phenyl ketone (2.07 mL, 15.0 mmol) was dissolved in MeOH (75 mL) and cooled in an icebath. Then, NaBH4 (851 mg, 22.5 mmol) was added in portions and the reaction mixture was stirred at roomtemperature for 12 hours. Then, the reaction mixture was quenched by NH4Cl aq. solution, and extracted withEt2O for three times. The combined organic layers were washed with brine, dried over Na2SO4, filtered througha plug of cotton, and concentrated under reduced pressure to give the corresponding alcohol S-22 which wasused in next step without further purification. To a solution of S-22 in MeCN/H2O (1/1, 100 mL) was addedp-TsOH?H2O (428 mg, 2.25 mmol). The mixture was stirred at 50 C for 27 h. The reaction was quenched withsat. NaHCO3 aq. at 0 C and extracted with AcOEt. The organic layer was washed with brine, dried overNa2SO4, filtered through a plug of cotton, and concentrated under reduced pressure. The residue was purifiedby flash column chromatography (AcOEt/Hexane = 1/4 to 1/3) to afford S-23 in 83% yield in 2 steps (1.86 g,12.6 mmol) as an amorphous. 1H NMR (400 MHz, CDCl3) δ: 1.50 (brs, 1H), 2.49 (dt, J = 6.0, 6.4 Hz, 2H),3.76 (dt, J = 5.2, 5.6 Hz, 2H), 6.21 (dt, J = 7.2, 15.6 Hz, 1 H), 6.50 (d, J = 16.0 Hz, 1H), 7.22 (dd, J = 6.8, 7.2Hz, 1H), 7.30 (dd, J = 7.2, 7.6 Hz, 2H), 7.36 (d, J = 7.6 Hz, 2H). Spectral data were identical to the reporteddata. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping