| Identification | Back Directory | [Name]
Plx-4032 (RG7024) | [CAS]
1029872-54-5 | [Synonyms]
RG 7204
Zelboraf
RO 5185426
vemurafenib
Plx-4032 (RG7024)
Vemurafenib, >=98%
VeMurafenib,PLX 4032
Plx-4032 (RG7024) USP/EP/BP
PLX 4032; RG 7204; RO 5185426
Vemurafenib Plx-4032 (RG7024)
PLX4032,Vemurafenib, Free Base
-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-B]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonaMide
N-[3-[[5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]carbonyl]-2,4-difluorophenyl]-1-PropanesulfonaMide
N-(3-{[5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]carbonyl}-2,4-difluorophenyl)propane-1-sulfonaMide
1-PropanesulfonaMide, N-[3-[[5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]carbonyl]-2,4-difluorophenyl]-
Propane-1-sulfonic acid {3-[5-(4-chloro-phenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl]-2,4-difluoro-phenyl}-amide
(1R)-1-(4-Hydroxy-3-{[(1R)-6-methoxy-1-(4-methoxybenzyl)-2-methyl -1,2,3,4-tetrahydro-7-isoquinolinyl]oxy}benzyl)-6-methoxy-2-methy l-1,2,3,4-tetrahydro-7-isoquinolinol | [EINECS(EC#)]
800-227-2 | [Molecular Formula]
C23H18ClF2N3O3S | [MDL Number]
MFCD18074504 | [MOL File]
1029872-54-5.mol | [Molecular Weight]
489.922 |
| Hazard Information | Back Directory | [Uses]
Vemurafenib selective BRAFV600E kinase inhibitor; an antitumor agent. Vemurafenib functions by inhibiting the proliferation and mitogen-activated protein/extracellular signal-regulated kinase (ERK) kinase and ERK phosphorylation in a panel of tumor cell lines, including melanoma cell lines expressing BRAFV600E or other mutant BRAF proteins altered at codon 600. Potent B-Raf inhibitor | [Definition]
ChEBI: Vemurafenib is a pyrrolopyridine that is 1H-pyrrolo[2,3-b]pyridine which is substituted at position 5 by a p-chlorophenyl group and at positions 3 by a 3-amino-2,6-difluorobenzoyl group, the amino group of which has undergone formal condensation with propane-1-sulfonic acid to give the corresponding sulfonamide. An inhibitor of BRAF and other kinases. It has a role as an antineoplastic agent and a B-Raf inhibitor. It is a pyrrolopyridine, a sulfonamide, a member of monochlorobenzenes, a difluorobenzene and an aromatic ketone. | [Brand name]
Zelboraf | [General Description]
Class: dual threonine/tyrosine kinase;
Treatment: melanoma with BRAF mutations; Elimination half-life = 57 h;
Protein binding > 99% | [Pharmacokinetics]
1 | [Pharmacokinetics]
The recommended daily dose of vemurafenib is
1,920 mg (4 × 240 mg tablets, BID), the highest
dosage among the three FDA-approved RAF
inhibitors (Table 2). One contributing factor for such
a high dose is the poor and variable oral
bioavailability due to both low cell permeability and
poor aqueous solubility. Nevertheless, vemurafenib
is absorbed rapidly after a single oral dose of 960 mg,
reaching a maximum drug concentration
approximately 4 h after administration. It also
exhibits long elimination half-life (57 h). It is cleared
predominantly via the hepatic route. Following oral administration, the parent drug
predominates in the plasma, and the isomeric
monohydroxylated species 5 are the only
metabolites detected in the plasma due to CYP3A4
mediated oxidation (Fig. 6).
  | [Clinical Use]
Vemurafenib was originally discovered at Plexxikon and has
been co-developed by Roche and Plexxikon as an oral BRAF
inhibitor for the treatment of patients with BRAFV600E mutation-
positive metastatic melanoma. The drug displays good potency
and selectivity for the V600E mutation (IC50 = 3.2–14 nM), an
oncoprotein, over the wild-type BRAF (IC50 = 21–370 nM). The
compound is less potent in in vitro kinase assays than other Plexxikon
BRAF inhibitors, but it was selected for clinical development based on its enhanced potency against the BARFV600E-containing
A374 melanoma cell line. | [Synthesis]
The synthesis described below is
based on a recent process patent (the Scheme).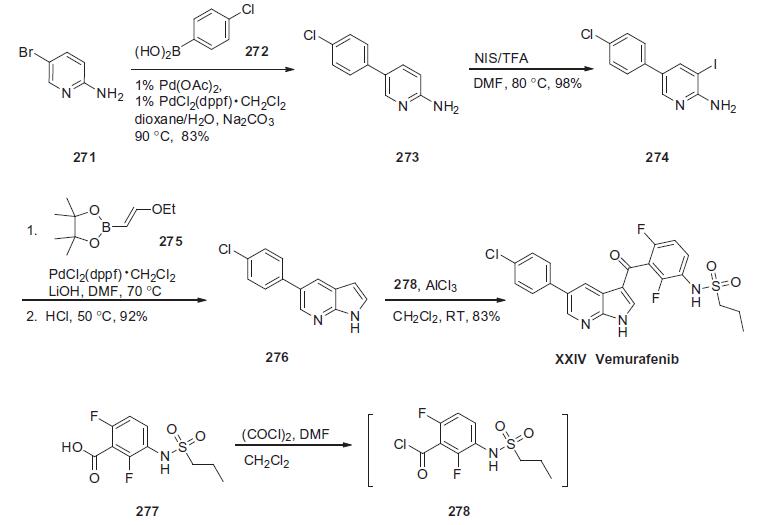
Commercially available 2-amino-5-bromopyridine (271) was
treated with 4-chlorophenylboronic acid (272) in the presence of
Na2CO3 and a catalytic amount of Pd(OAc)2/PdCl2(dppf)�CH2Cl2 to
give Suzuki product 273 in 83% yield. Arene 273 was subjected
to iodination conditions using NIS and TFA to provide iodide 274
in 98% yield. Iodide 274 and pinacol vinylboronate 275 were coupled
under Suzuki conditions followed by treatment with acid to
affect a tandem coupling¨Ccyclization sequence which resulted in
pyrimidyl pyrrole 276 in good yield. This material was treated with
aluminum trichloride and then subjected to the the acyl chloride of
commercially available sulfonamide acid 277, triggering a Friedel-
Crafts reaction providing vemurafenib (XXIV) in 85% yield. | [target]
Primary target: BRAF | [Drug interactions]
Potentially hazardous interactions with other drugs
Anticoagulants: possibly enhances anticoagulant
effect of warfarin.
Antipsychotics: avoid concomitant use with
clozapine, risk of agranulocytosis.
Oestrogens and progestogens: contraceptive effect
possibly reduced. | [Metabolism]
Only 5
% of a dose of vemurafenib is metabolised. 94
% of the dose is excreted in the faeces and 1
% in the urine. |
|
|